
HYDRANENCEPHALY IN PUBLIC HOSPITAL OF QUINTANA ROO. CASE SERIES
Renán Baqueiro-Canto1, Guillermo Padrón-Arredondo1
1 Hospital General de Playa del Carmen. Servicios Estatales de Salud de Quintana Roo. México.
Correspondencia: Renán Baqueiro-Canto. Correo electrónico: Esta dirección de correo electrónico está protegida contra spambots. Usted necesita tener Javascript activado para poder verla. Esta dirección de correo electrónico está protegida contra spambots. Usted necesita tener Javascript activado para poder verla.
Recibido: 23 de abril de 2019.
Aceptado: 24 de mayo de 2019.
RESUMEN
Introducción. Varios tipos de encéfalo-displasias asociadas con formaciones quísticas han sido descritas. En 1904 Thurnbull describió un caso bajo el término de aplasia quística, en donde ambos lóbulos parietales estaban involucrados. En 1950 Hamby, Krauss y Beswick reportaron siete niños con displasia utilizando el término hidranencefalia, como es conocida actualmente.
Caso clínico 1. Paciente neonato masculino de término obtenido por cesárea, con Apgar, Silverman y somatometría normales, de madre de 23 años de edad, portadora de VIH sin control prenatal a quien al hacer ultrasonido obstétrico se identificó como hidranencefalia vs ventriculomegalia obstructiva severa. El ultrasonido y la tomografía comprueban el diagnóstico. El paciente fallece a los siete días de su nacimiento.
Caso clínico 2. Masculino de tres meses de edad, producto del segundo embarazo de 35 semanas de gestación por parto vaginal que no respira al nacer y es intubado; la ecografía obstétrica identificó hidrocefalia. El nuevo ultrasonido transfontanelar mostró hidranencefalia pendiente de resonancia magnética nuclear para reconfirmación diagnóstica. El diagnóstico definitivo por ultrasonido transfontanelar fue hidranencefalia.
Discusión. La etiología de la hidranencefalia es controversial, pero ya que las meninges y el cráneo están intactos, se presume que desde los rudimentos del neopallium el defecto ya está presente durante el periodo de formación embriológica de estas capas. Los datos de neuroimagen fetal y postnatal y los hallazgos histopatológicos apuntan hacia una oclusión precoz de las arterias carótidas internas, que se presenta principalmente entre la octava y la duodécima semana de gestación, como el principal mecanismo patogénico.
Palabras clave: Hidranencefalia; Arterias Carótidas; Diagnóstico Prenatal; Diagnóstico Postnatal.
ABSTRACT
Introduction. Several types of encephalo-dysplasias associated with cystic formations have been described. In 1904 Thurnbull described a case under the term cystic aplasia, where both parietal lobes were involved. In 1950 Hamby, Krauss and Beswick reported seven children with dysplasia using the term hydranencephaly, as it is known today.
Clinical case 1. Term male newborn patient obtained by caesarean section with Apgar, Silverman and normal somatometry, from a mother of 23 years of age, HIV carrier without prenatal control who was identified by obstetric ultrasound as hydranencephaly vs severe obstructive ventriculomegaly. Ultrasound and CT scan prove the diagnosis. The patient dies seven days after birth.
Clinical case 2. Three-month-old male, product of the second pregnancy of 35 weeks gestation by vaginal delivery that does not breathe at birth and is intubated; obstetric ultrasound identified hydrocephaly. The new transfontanellar ultrasound showed hydroencephaly pending nuclear magnetic resonance for diagnostic reconfirmation. The definitive diagnosis by transfontanellar ultrasound was hydroencephaly.
Discussión. The aetiology of hydranencephaly is controversial but since the meninges and skull are intact, it is presumed that from the rudiments of neopallium the defect is already present during the period of embryological formation of these layers. Fetal and postnatal neuroimaging data and histopathological findings point to early occlusion of the internal carotid arteries, which occurs mainly between the eighth and twelfth week of gestation, as the main pathogenic mechanism.
Key words: Hydranencephaly; Carotid Arteries; Prenatal Diagnosis; Postnatal Diagnosis.
INTRODUCCIÓN
Varios tipos de encéfalo-displasias han sido descritas asociadas con formaciones quísticas. Thurnbull en 1904 describió un caso bajo el término de aplasia quística, en donde ambos lóbulos parietales estaban involucrados. En 1950 Hamby, Krauss y Beswick reportaron siete niños con displasia, utilizando el término hidranencefalia, como es conocida actualmente. Esta condición también fue descrita en 1940 por Bettinger y por primera vez por Cruveilhier en 1835. El más antiguo caso corresponde a Ambroise Paré (1-3).
La hidranencefalia es una anomalía encefaloclástica caracterizada por la ausencia y reemplazo de los hemisferios cerebrales por líquido cefalorraquídeo y desechos necróticos, cubiertos por leptomeninges. Esta condición es el resultado de la destrucción y reabsorción de tejido cerebral sólido preformado que comienza antes del nacimiento (4).
El mecanismo patogénico actualmente aceptado es una obstrucción interna bilateral de la arteria carótida con evidencia de que el proceso podría iniciar desde 8-12 semanas de gestación (5). Partes del cerebro suministradas por las arterias vertebrales y cerebrales posteriores, como el cerebelo, el tronco del encéfalo, el tálamo y los ganglios basales, así como el plexo coroideo, generalmente son conservados.
Las complicaciones asociadas son variables como las oculares asociadas e incluyen anomalías pupilares, estrabismo, nistagmo, ptosis, hipoplasia del nervio óptico, coriorretinitis, estrechamiento de los vasos sanguíneos de la retina y síndrome de división de la cámara anterior; osteogénesis imperfecta; diabetes insípida y displasia renal, entre otras (6).
Caso 1
Paciente neonato masculino de término obtenido por cesárea con Apgar, Silverman y somatometría normales, de madre de 23 años de edad portadora de VIH, sin control prenatal a quien el ultrasonido obstétrico identificó hidranencefalia vs ventriculomegalia obstructiva severa. Un nuevo ultrasonido reportó: región supratentorial con quiste que ocupa el espacio superior. No se identifica línea media ni masa cerebral, solamente septo interno incompleto y plexos coroideos, tampoco es visible tálamo ni ganglios basales. Se observan estructuras de la base posterior y no hay flujo en doppler color ni en doppler dependiente (figuras 1 y 2). Se le solicitó tomografía que reporta huesos de cráneo y fontanelas abiertas. Cavidad quística que ocupa la totalidad del espacio cerebral, fosa pontina con estructura similar a reminiscencia cerebelosa izquierda y tallo cerebral (figuras 3 y 4). El paciente fallece a los siete días de su nacimiento en la unidad de cuidados intensivos neonatales.
Figura 1. Imagen ecográfica transfontanelar del caso 1, donde se observa imagen quística que abarca casi la totalidad del parénquima cerebral supratentorial.
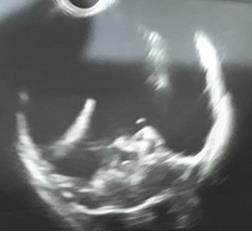
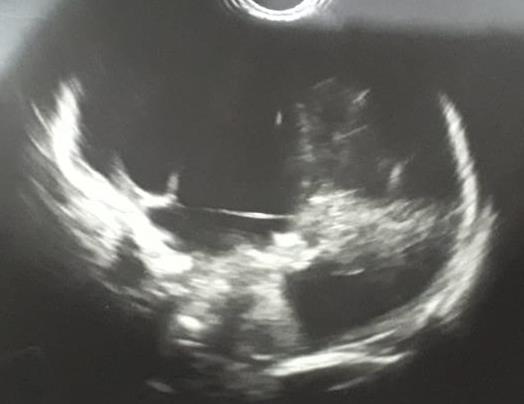
Figura 2. Tomografías sagital y coronal del caso 1, donde se observa escaso parénquima cerebral con predominio en región posterior e imagen quística que ocupa toda la región supratentorial que desplaza las estructuras superiores.
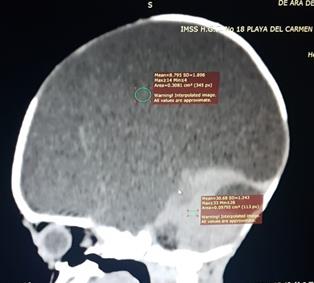
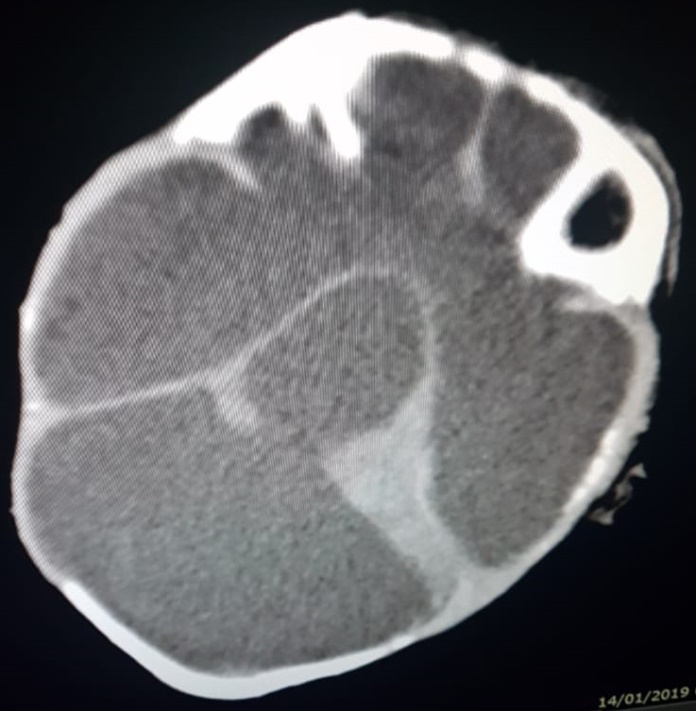
Figura 3. Ultrasonido transfontanelar del caso 2, donde se observa imagen quística que abarca casi la totalidad del parénquima cerebral.
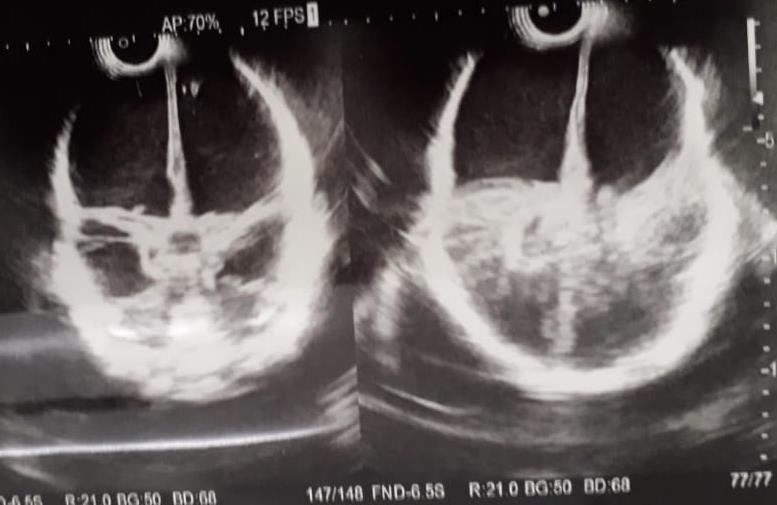
Caso 2.
Paciente masculino de tres meses de edad, producto del segundo embarazo, obtenido a las 35 semanas de gestación por parto vaginal que no respira al nacer y está intubado. Capurro de 35 semanas de gestación. Una madre de 20 años con antecedentes de infección urinaria y del tracto urinario recurrente que recibió fumarato ferroso y vitaminas de su chequeo prenatal. La ecografía obstétrica identificó hidrocefalia, se recomendó la interrupción del embarazo, que no fue aceptada. El nuevo ultrasonido transfontanelar mostró hidranencefalia pendiente de resonancia magnética nuclear para reconfirmación diagnóstica (Figuras 1 y 2). La paciente tiene actualmente tres meses de edad con diagnóstico clínico de hidrocefalia congénita, convulsiones recurrentes y anemia. Y el diagnóstico definitivo por ultrasonido transfontanelar es el de hidranencefalia.
DISCUSIÓN
La etiología de la hidranencefalia es controversial, pero ya que las meninges y el cráneo están intactos, se presume que desde los rudimentos del neopallium el defecto ya está presente durante el periodo de formación embriológica de estas estructuras. (6)
Por otra parte, la hidranencefalia es una malformación poco frecuente, con una incidencia aproximada de 1 en 10 000 nacidos vivos; se caracteriza por la ausencia de hemisferios cerebrales, los cuales son reemplazados por un saco membranoso que contiene líquido cefaloespinal. El origen de esta patología se desconoce. La explicación más probable para la desaparición de los hemisferios cerebrales fetales es la oclusión de la porción supraclinoidea de ambas arterias carótidas (se han encontrado, en estudios post natales, arterias hipoplásicas o aplasia bilateral con la consiguiente destrucción hemorrágica del parénquima cerebral (7).
La detección variable de restos cerebrales parece reflejar la vía de desarrollo de las arterias cerebrales. Además, los datos de neuroimagen fetal y postnatal y los hallazgos histopatológicos apuntan hacia una oclusión precoz de las arterias carótidas internas, que se presenta principalmente entre la octava y la duodécima semana de gestación, como el principal mecanismo patogénico (8). De tal manera que es un reto para los imagenólogos detectar esta anomalía en etapas tempranas del desarrollo fetal.
Por otra parte, su etiología ha sido atribuida a múltiples causas que conducen a lesión isquémica y las infecciones virales son las principales implicadas, como herpes virus, parvovirus, citomegalovirus, entre otros; otra posible etiología son los procesos tóxicos (alcohol) o genéticos como por ejemplo síndrome de Fowler (9, 10). La hidranencefalia no se presenta posterior a una hidrocefalia, mientras que sí puede iniciarse como encefalopatía multiquística, y progresar a hidranencefalia, ya que en ésta también hay una lesión vascular severa de base (11).
También se ha informado de casos de hidranencefalia asociados con toxoplasmosis congénita como otra posibilidad etiológica de este padecimiento, aunque en estos casos, lo más probable es la hidrocefalia (12) así como asociada con lupus eritematoso sistémico (13).
La genética también se ha abocado a desentrañar este misterio, de tal manera que Abdel-Hamid MS, et al. (14), los llevó a considerar la nudE [neurodevelopment protein 1 gene (NDE1)] por sus siglas en inglés, e identificar una nueva variante no sensible homocigótica (c.54G> A, p.W18) como posible causa de la enfermedad. La variabilidad del grado de malformaciones cerebrales y la fusión aparente del tálami fueron elusivas y retrasaron el reconocimiento de la etiología genética. Sus resultados proporcionan la primera descripción prenatal de este raro síndrome.
En cuanto al diagnóstico por imágenes, Chih-Ping Chen, et al (15), informan de un caso con displasia tanatofórica tipo II, detectada mediante ultrasonido prenatal, observando translucencia nucal a las 14 semanas de gestación y un nuevo estudio a las 25 semanas reveló hidranencefalia asociada con otros defectos. Kline-Fath BM, et al. (14) refieren que la ventriculomegalia fetal es una referencia común para la RM prenatal con posibles etiologías como hidrocefalia e hidranencefalia.
Gardea-Loera G, et al. (16) discuten los aspectos clínicos de neuroimagen y comportamiento electrofisiológico de la hidranencefalia donde existe gran similitud entre hidranencefalia e hidrocefalia congénita severa, tanto clínicamente como en estudios de imagen. En la hidranencefalia, habitualmente el electroencefalograma (EEG) no muestra actividad eléctrica y los potenciales evocados auditivos del tallo cerebral son normales.
CONCLUSIÓN
En nuestro hospital llama la atención la presentación de estos dos casos en tan corto tiempo y además son los primeros casos que se presentan en nuestra comunidad. Actualmente la etiología definitiva de la hidranencefalia aún no se ha podido establecer y se requiere estar más atentos para detectar más casos y así poder establecer una correlación más precisa.
REFERENCIAS
1. Hubert M, Turnbull MA. Bilateral loss of postcentral cortex, apparently congenital, in an adult. Brain 1904; 27(2):209-51.
2. Watson KC. Hydranencephaly; clinical diagnosis; presentation of 7 cases. Arch Dis Child 1956 Jun; 31(157):195-7.
3. Hino-Fukuyo N, Togashi N, Takahashi R, Saito J, Inui T, Endo W, et al. Neuroepidemiology of Porencephaly, Schizencephaly, and Hydranencephalyin Miyagi Prefecture, Japan. Pediatr Neurol2016 Jan; 54:39-42.
4. Crome L, Sylvester PE. Hydranencephaly (Hydrencephaly). Arch Dis Child1958; 2013:235-45.
5. Cecchetto G, Milanese L, Giordano R, Viero A, Suma V, Manara R. Looking at the missing brain: hydranencephaly case series and literature review. Pediatr Neurol2013 Feb; 48(2):152-8.
6. Hamby WB, Krauss RF, Beswick WF. Hydranencephaly; Clinical diagnosis; presentation of 7 cases. Pediatrics 1950 Sep; 6(3):371-83.
7. Lacunza Paredes RO, Correa López W. Hidranencefalia como presentación más severa de apoplejía cerebral fetal: a propósito de dos casos. Rev Peruana Ginecol 2014; 60(2): 183-87.
8. Fowler M, Dow R, White TA, Greer CH. Congenital hydrocephalus-hydrocephaly in five siblings, with autopsy studies: a new disease. Develop Med Child Neurol 1972; 14.173-88.
9. Radio FC, Di Meglio L, Agolini E, Bellacchio E, Rinelli M, Toscano P, et al. Proliferative vasculopathy and hydranencephaly–hydrocephaly syndrome or Fowler syndrome: Report of a family and insight into the disease’s mechanism. Mol Genet Genomic Med 2018; 6:446-51.
10. Castillo AM, Mena Olmedo G, Hernández D, Carrillo E, Aguirre J. Diagnóstico intraútero de hidranencefalia en relación a un caso clínico. www.webcir.org/revistavirtual/articulos/junio14/ecuador/ecu_esp. 6 págs.
11. Gaete MB, Estay NA, Mesa LT. Hidranencefalia en un recién nacido por toxoplasmosis congénita. Rev Chil Pediat 2011; 82(5):19-425.
12. McAdams RM. Maternal systemic lupus erythematosus and hydranencephaly in a neonate: a case report. J Matern Fetal Neonat Med 2005; 18:279-81.
13. Abdel-Hamid MS, El-Dessouky SH, Ateya MI, Gaafar HM, Abdel-Salam GMH. Phenotypic spectrum of NDE1-related disorders: from microlissencephaly to microhydranencephaly. Am J Med Genet A2019 Jan 13. doi:10.1002/ajmg.a.61035.
14. Chih-Ping Chen, Tung-Yao Chang h , Tan-Wei Lin h , Schu-Rern Chern , Shin-Wen Chen, Shih-Ting Lai, et al. Prenatal diagnosis of hydrancephaly and enlarged cerebellum and cisterna magna in a fetus with thanatophoric dysplasia type II and a review of prenatal diagnosis of brain anomalies associated with thanatophoric dysplasia. Taiwanese J Obstet Gynecol 2018; 57:119-22.
15. Kline-Fath BM, Merrow AC Jr, Calvo-Garcia MA, Nagaraj UD, Saal HM. Fowler syndrome and fetal MRI findings: a genetic disorder mimicking hydranencephaly/hydrocephalus. Pediatr Radiol.2018 Jul; 48(7):1032-34.
16. Gardea-Loera Gilberto, Velazco-Campos M. Aspectos clínicos de neuroimagen y comportamiento electrofisiológico de la hidranencefalia. Arch Neurocien (Mex) INNN 2014; 19(1) enero-marzo: 48-52.




