
Síndrome por deleción en el cromosoma 18q. Caso clínico
Deletion Syndrome in Chromosome 18q. Case report
Autores: Alejandro Gaviño Vergara, Carlos Alonso Muñoz, Carlos Cortes Penagos, Karla Nathalie Gaytán Nares
Sede: CRIT Quintana Roo y Laboratorios Mendel, Universidad Michoacana de San Nicolás de Hidalgo.
Correspondencia: Dr. Alejandro Gaviño Vergara. CRIT Cancún Blv. Luis Donaldo Colosio 999 Smz. 296 Col. Alfredo V. Bonfil, Can Cún Quintana Roo, México. CP: 77560
Correo electrónico: GaviñEsta dirección de correo electrónico está protegida contra spambots. Usted necesita tener Javascript activado para poder verla.
RESUMEN
El síndrome por deleción 18q o también llamado monosomía parcial del 18q, es una entidad de causa genética con una incidencia de 1 /55 000 nacimientos, predomina en el sexo femenino y es incluido dentro de la clasificación de las enfermedades raras. Las principales características de este síndrome son dismorfias faciales, talla baja, pérdida auditiva, hipotonía, retraso mental, deformación en pies, alteraciones endócrinas y autoinmunes. El diagnóstico se puede realizar por medio del estudio citogenético de cariotipo en sangre periférica, sin embargo esto dependerá del tamaño de la región involucrada. El diagnóstico oportuno de esta patología y el abordaje multidisciplinario de estos pacientes podrá dar una mayor y mejor calidad de vida a ellos y sus familias.
Palabras clave: deleción 18q, cariotipo, calidad de vida.
ABSTRACT
18q deletion syndrome or also called partial 18q monosomy is a genetic condition with an incidence of 1 / 55,000 births, it predominates in females and it’s included with the classification of rare diseases. The main characteristics of this syndrome are facial dysmorphism, short stature, hearing loss, hypotonía, mental retardation, foot deformity, endocrine and autoimmune disorders. The diagnosis can be made through cytogenetic study of blood karyotype, however this will depend on the size of the region involved. The timely diagnosis of this pathology and the multidisciplinary approach of these patients will be able to give a greater and better quality of life to them and their families.
Keywords: 18q deletion, karyotype, quality of life.
INTRODUCCION
El síndrome 18q-, también denominado monosomía parcial 18q, fue inicialmente descrito por Grouchy en 1964(1).
Es un síndrome de causa genética, con una incidencia de 1 /55 000 nacimientos, predomina en el sexo femenino y es incluido dentro de la clasificación de las enfermedades raras(2) sin embargo, es el segundo síndrome más común que involucra al cromosoma 18(3,4).
Cerca del 94% de los casos que cursa con la deleción 18q son casos de novo, y el 6% restante son heredados por alguno de los padres, los cuales pueden presentar una translocación cromosómica balanceada(5).
El síndrome 18q- presenta una alta variabilidad fenotípica, (6) lo que hace difícil su diagnóstico clínico y a su vez provoca que un niño con la deleción parcial 18q- pueda tener trastornos en su desarrollo, de forma global, diferente a otro niño con el mismo diagnóstico.
Las principales características de este síndrome son dismorfias faciales, talla baja, pérdida auditiva, hipotonía, retraso mental, deformación en pies, alteraciones endócrinas y autoinmunes(7).
CASO CLÍNICO
Paciente femenino de 2 años 4 meses de edad, cuenta con los siguientes antecedentes papá de 34 años al nacimiento originario de Quintana Roo, mamá de 28 años originaria de Puebla, consanguinidad negada. Hermana de 4 años sana. No hay antecedente familiares relacionados a patología de base.
Producto de la gesta 3, cesárea 1, aborto 1, control prenatal a partir de la 3ª semana, ingesta de ácido fólico, al 5º mes progesterona hasta el nacimiento por amenaza de parto prematuro, ultrasonidos mensuales normales. Obtenida a las 38.5 semanas por cesárea programada, peso 3 kg, talla 49 cm, Apgar 9-9, egresa como sana.
Desarrollo psicomotor: Motor grueso y fino: control de cuello 6 meses, tronco 8 meses, bipedestación 18 meses, comienza a intentar la marcha. Lenguaje: balbuceo a los 3 meses, actualmente 2 años emite sonidos monosilábicos, no logra bisílabos. Persona social: sonrisa social 6/12,
Estudios laboratorio y gabinete: Electroencefalograma. Reportado normal; Ultrasonido abdominal sin alteraciones; Ecocardiograma: Reporta comunicación interauricular ostium secundum de 6.9x7x5 mm, presión pulmonar normal; Tamiz auditivo: reportan hipoacusia profunda bilateral; PEA: hipoacusia derecha severa a profunda, izquierda severa; TAC de oídos que reporta disgenesia de membranas timpánicas y martillo. Impedanciometría otitis media serosa bilateral
Padecimiento actual: Al nacimiento debido a pie valgo acuden a consulta médica le refieren que era posicional y que cedería con el tiempo, por otro lado notaban que era poco reactiva al medio, acudieron con neuropediatra encontrando hipotonía y retraso psicomotor, sugirió terapia física con lo que tuvo poco mejoría, fue valorada por el servicio de Genética el cual debido a dismorfias faciales y retraso en el neurodesarrollo solicito cariotipo en sangre periférica con el resultado: 46,XX,del(18)(q21.3) en 20 metafases analizadas. (Fig. 1)
Figura 1. Cariotipo en sangre periférica donde señala (flecha roja) la deleción del brazo largo del cromosoma 18.

Exploración física: Peso: 10 kg (debajo percentila 3) Talla: 79 cm. (debajo percentila 3) Perímetro cefálico (PC): 44cm. (debajo percentila 3); signos vitales: Saturación de oxígeno (Spo2 T): 98% Frecuencia cardiaca (FC): 99 x min. Frecuencia respiratoria (FR): 23 x min. Tensión arterial (T/A): 80/60 mm de Hg. Temperatura: 36.3°C. Pulso: 99 x min. Dismorfias faciales como epicanto bilateral, puente nasal deprimido, pabellones auriculares baja implantación, ruidos cardiacos sin alteraciones, abdomen sin hernias ni megalias, hiperlaxitud ligamentaria de predominio en miembros inferiores, dorso con hoyuelo presacro.
DISCUSION
La importancia de presentar este caso radica en la sospecha diagnóstica desde los primeros meses de vida al encontrar su médico retraso en su desarrollo así como rasgos faciales los cuales nos pueden hacer pensar en una entidad cromosómica. Debido a esto fue enviado a Genética y al realizar el cariotipo se confirmó el diagnóstico de deleción cromosómica 18.
Las características reportadas en la literatura comparadas con la del paciente son las siguientes:
A los 10 meses de edad observan cara redonda cejas dispesas, epicanto,puente nasal deprimido punta nariz bulbosa.

Un año 8 meses de edad con fisuras palpebrales oblicuas hacia arriba, epicanto, puente nasal deprimido, punta nariz bulbosa, comisura labial hacia abajo.
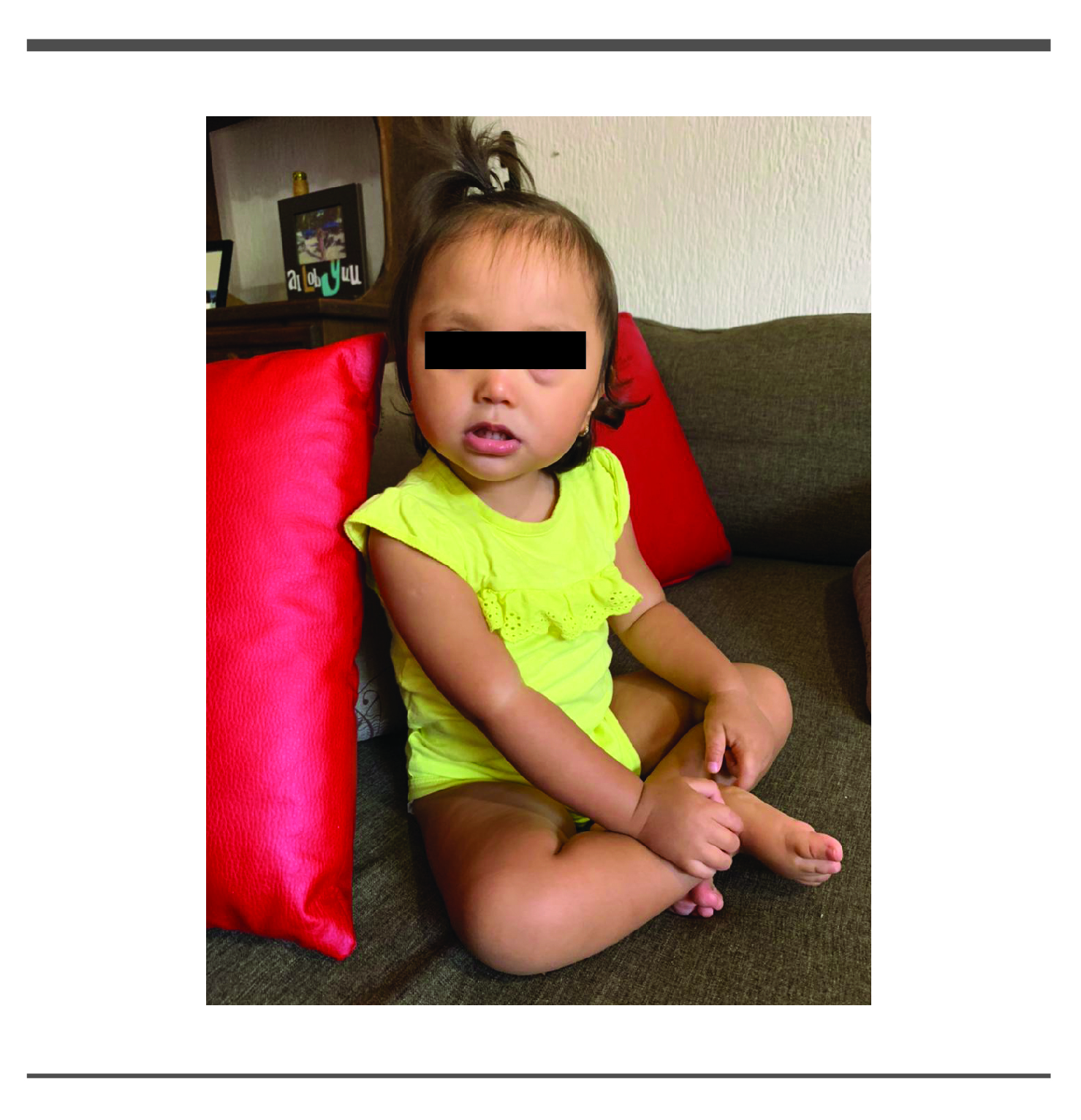
Paciente edad actual 2 años 4 meses.

En la literatura se refiere que en la resonancia magnética cerebral pueden presentar cambios en la sustancia blanca motivo por el cual se buscará intencionadamente este hallazgo.
Podemos observar que los datos clínicos que se reportan en la deleción 18 q los comparte nuestra paciente con excepción de los defectos en paladar.
En nuestra paciente tiene una deleción de 18q21 hasta la región terminal del cromosoma 18 datos que coinciden con lo reportado en la literatura.
El diagnosticar esta patología durante los primeros meses de vida, nos permite darle una mejor calidad de vida al paciente atendiendo los problemas motores desde inicio temprano, terapia del lenguaje y uso de auxiliares auditivos así como seguimiento por Neurología por el riesgo de desarrollo crisis convulsivas.
CONCLUSIONES
El síndrome de deleción 18q es una condición muy heterogénea con un fenotipo muy variable, generalmente caracterizado por retraso mental, baja estatura, hipotonía, deterioro de la audición, características dismórficas, niveles bajos de inmunoglobulina A y deficiencia de la hormona del crecimiento. La atención temprana en estos pacientes podrá mejorar sin duda alguna, su calidad de vida al igual que la de sus familiares.
El autor no declara conflicto de intereses
Este trabajo no tuvo patrocinadores.
REFERENCIAS
- Loevner LA, Shapiro RM, Grossman RI, Overhauser J, Kamholz J. White matter changes associated with deletions of the long arm of chromosome 18 (18q2 syndrome): a dysmyelinating disorder? AJNR [Internet]. 1996 Nov [citado 9 Ene 2019]; 17:1843-8.
- Larner AJ. Deletion of 18q. En: DADS. Reference Work Entry; 2009. p. 503-4 [citado 9 Ene 2019].
- Córdova-Fletes C, Sainz-González E, Avendaño-Gálvez RI, Ramírez-Velazco A, Rivera H, Ortiz-López R, et al. De novo dir dup/del of 18q characterized by SNP arrays and FISH in a girl child with mixed phenotypes. J Genet [Internet]. 2014 Dec [citado 9 Ene 2019]; 93(3):869–73.
- Chen H. R (18) Syndrome. Atlas of Genetic Diagnosis and Counseling [Internet]. Springer Science Business Media LLC; 2017. p. 2417-25 [citado 9 Ene 2019].
5. Elif Ozsu, Yesiltepe Mutlu, Butel Aysegul. Features of two cases with 18q deletion syndrome, J Clin Res Pediatr Endocrinol. 2014; 6(1):51-4
6. Bohiltea RE, Cirstoiu MM, Nedelea FM, Turcan N, Giorgescu TA, Munteanu O, et al. Case report of a novel phenotype in 18q deletion syndrome, Rom J Morphol Embryol. 2020 Jul-Sep; 61(3):905-910.
7. Budisteanu M, Arghir A, Chirieac, Sorina M, Tutulan-Cunita A, Lungeanu Agripina. 18q deletion syndrome - A case report, Mædica - J Clin Med. 2010; 5(2); 135-8.



