
Estatus epiléptico súper-refractario secundario a hipoglicemia en paciente pediátrico: Caso clínico
Super-refractory status epilepticus secondary to hypoglycemia in a pediatric patient: Case report.
Autores: Paula Ireri Pech-Marín, Antonio Azael Couoh-Poot, Dennis Alexander Sánchez-Marrufo, Diana Sofía Estrella-Olguín.
.Sede: Hospital General de Chetumal, Q. Roo
Correspondencia:Paula Ireri Pech-Marín
Correo electrónico: Esta dirección de correo electrónico está protegida contra spambots. Usted necesita tener Javascript activado para poder verla.
Recibido: 23 de mayo del 201
Aceptado: 20 de noviembre del 2019
RESUMEN
Introducción. El estatus epiléptico súper-refractario (EESR) es una emergencia neurológica rara en pediatría con alto índice de morbi-mortalidad con una amplia variedad de etiologías. Caso clínico. Se presenta el caso de una paciente femenina de 2 años en EESR secundario a hipoglicemia la cual persistió con crisis convulsivas tras manejo con dos líneas de anticomiciales e inducción anestésica con barbitúricos. Discusión.En la actualidad no se cuenta con guías específicas para el manejo del EESR, basándose únicamente en la opinión experta o reporte de casos. Broomall et al describieron dos casos de pacientes pediátricos en EESR manejados con allopregnalona, los cuales presentaron mejoría de las crisis tras su administración, pero requirieron continuar con manejo de tercera línea por ciertos periodos de tiempo. Algunos autores sugieren el uso de ketamina e hipotermia; otras sugerencias incluyen terapia inmunológica, dieta cetogénica, administración de piridoxina y neurocirugía. De igual manera se recomienda mantener la infusión de antiepilépticos de forma continua. Otras terapias emergentes incluyen la terapia electroconvulsiva y la estimulación magnética transcraneal; sin embargo, existe poca información sobre la eficacia y seguridad de estas terapias, por lo que sería recomendable ampliar los estudios sobre el manejo del EESR. Conclusiones.A pesar de no contar con protocolos establecidos para el manejo del EESR, el reconocimiento de la etiología y el inicio del tratamiento no deben de retrasarse para disminuir riesgo de morbi-mortalidad y complicaciones neurológicas y sistémicas.
Palabras clave: pediatría; Estatus epiléptico; Estatus epiléptico súper-refractario; Hipoglicemia.
ABSTRACT
Introduction. The super-refractory status epilepticus (SRSE) is a rare neurological emergency in pediatrics with a high morbidity and mortality rate and a wide variety of etiologies. Clinical case. We report the case of a 2-year-old female patient in SRSE secondary to hypoglycemia who persisted with seizures after management with two lines of anticonvulsants and anesthetic induction with barbiturates. Discusion. At present, there are no specific guidelines for the management of EESR, based solely on expert opinion or case reports (5). Broomall et al described two cases of pediatric patients in SRES managed with allopregnalone, which presented improvement of the seizures after its administration, but required to continue with third-line management for certain periods of time. Some authors suggest the use of ketamine and hypothermia; other suggestions include immune therapy, ketogenic diet, administration of pyridoxine, and neurosurgery. Similarly, it is recommended to maintain the infusion of antiepileptic drugs continuously. Other emerging therapies include electroconvulsive therapy and transcraneal magnetic stimulation; however, there is little information on the efficacy and safety of these therapies, so it would be advisable to expand the studies on the management of RES. Conclusions. Despite not having established protocols for the management of SRSE, the recognition of the etiology and the initiation of treatment should not be delayed reducing the risk of morbidity and mortality, as well as neurological and systemic complications.
Keywords: Status epilepticus; Super-refractory status epilepticus; Pediatrics; Hypoglycemia.
INTRODUCCIÓN
Una crisis epiléptica se define como una emergencia médica provocada por una actividad neuronal anormal, excesiva e hipersincrónica del encéfalo, que puede ser desencadenada por trastornos metabólicos, infecciosos, inmunológicos, malformaciones congénitas, lesiones adquiridas a nivel de sistema nervioso central (SNC) o desconocidos (1).
El estatus epiléptico (EE) se define comúnmente como 5 minutos o más de crisis convulsivas o dos o más crisis sin recuperación del estado de consciencia (2,3). Este estado tiene una frecuencia de aproximadamente 20 episodios por 100,000 niños por año, con mortalidad de 4% en los casos de EE refractario al tratamiento, con retraso en la terapia inicial llevando a mayor duración de las crisis, mayor mortalidad, necesidad de infusiones continuas y aumento en la probabilidad de hipotensión (2).
El estatus epiléptico refractario (EER) se define como las convulsiones que no responden a medicamentos antiepilépticos de primera o segunda línea, con duración mayor a 1 hora o que requieren de anestesia general para su control (4,5). Una nueva subdefinición del mismo es el estatus epiléptico súper-refractario (EESR), definido como crisis convulsivas que se repiten 24 horas o más tras inducción de terapia anestésica, incluidas las crisis durante la reducción o retiro de la anestesia (6,7). Este ocurre entre 10 y 15% de los casos que se presentan en el hospital, con una tasa de mortalidad que varía entre 30 y 50% (6,8).
CASO CLÍNICO
Femenina de 2 años, originaria de Escárcega, Campeche y residente del poblado Niños Héroes, Quintana Roo. Antecedente de asma bronquial diagnosticada hace 6 meses, refiere una hospitalización por crisis asmática, actualmente en tratamiento con salbutamol. Niega antecedentes quirúrgicos, traumáticos, transfusionales y alérgicos. Esquema de vacunación aparentemente completo. Antecedentes familiares de importancia: madre portadora de diabetes mellitus tipo 2 en tratamiento con metformina y glibenclamida.
Inicia padecimiento el viernes 22 de marzo de 2019 aproximadamente a las 05:00 h, tras aparente ingestión accidental de desconocido número de tabletas de glibenclamida y metformina, con presencia de astenia, irritabilidad y llanto. Posteriormente presenta seis episodios de crisis convulsivas tónico-clónicas, sin recuperación del estado de consciencia, motivo por el cual es llevada a centro de salud del poblado Niños Héroes. Se realizan estudios de laboratorio que reportan: glucosa, 12.9 mg/dl; urea, 18.8 mg/dl; BUN 8.78 mg/dl; creatinina, 0.44 mg/dl; ácido úrico, 3.96 mg/dl; colesterol, 147 mg/dl; triglicéridos, 82.3 mg/dl; sodio, 136.2 mmol/L; potasio, 4.36 mmol/L; cloro, 102.7 mmol/L. Se yugulan crisis con diazepam y difenilhidantoína (DFH), se desconocen dosis manejadas, al igual de si se realizó infusión de glucosa. No obstante, las crisis convulsivas persisten, motivo por el cual es referida al Hospital General de Chetumal.
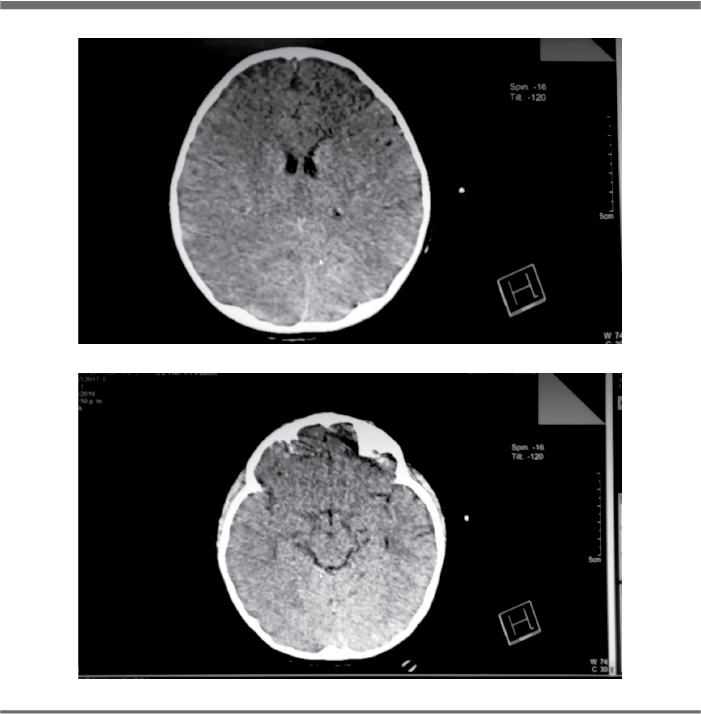
Se recibe a paciente bajo efectos de sedación, pupilas isocóricas con pobre respuesta a estímulo luminoso, faringe hiperémica con descarga retrofaríngea, campos pulmonares bien ventilados, precordio con taquicardia, sin ruidos agregados, abdomen blando, depresible, no doloroso, peristalsis disminuida; se reporta glicemia capilar de 56 mg/dl. Se inicia manejo mediante diazepam, levetiracetam, DFH e infusión con solución glucosada. Se realiza Tomografía Axial Computarizada (TAC) de cráneo la cual reporta datos de edema cerebral agudo (Fig. 1).
Figuras 1 A y B. TAC de cráneo simple donde se observa borramiento de las cisternas subaracnoideas y compresión de los surcos y giros cerebrales.
Pese al manejo con anticomiciales del día 23 al 25 de marzo, las crisis convulsivas no remiten, focalizándose en hemicara izquierda y miembro torácico ipsilateral de características mioclónicas, motivo por el cual se decide su ingreso a la Unidad de Cuidados Intensivos Pediátricos (UCIP), se realiza electroencefalograma (ECG) y se induce a un coma barbitúrico por Propofol y a ventilación mecánica asistida. ECG reporta «actividad theta y delta fronto-central y temporal bilateral sin observarse paroxismos; estado post-ictal probablemente metabólico». Se agrega infusión de solución hipertónica como medida angioedema.
El día 28 de marzo se decide el cambio de Propofol a fentanilo en infusión, y se inicia manejo con esteroide para valorar progresión ventilatoria, la cual se realiza sin complicaciones, manteniéndose con aporte de oxígeno con mascarilla reservorio. La paciente evoluciona con recurrencia de las crisis a pesar de manejo anticomicial y adecuado estado hemodinámico, respiratorio y nefro urológico, manejando valores normo glucémicos. El día 02 de abril se decide suspensión de DFH, agregándose topiramato por crisis convulsivas de difícil control. Debido a presentar adecuada saturación de oxígeno, se suspende fase II de ventilación progresándose a aire ambiente. Último episodio de crisis convulsivas reportado el 06 de abril.
El día 08 de abril, debido a no presentar nuevos episodios de convulsiones, se decide egreso de UCIP y pase a sala general del servicio de pediatría. Última valoración por neurocirugía reporta que TAC de cráneo presenta «zonas de isquemia a nivel bio-occipital, frontal y temporal derechos, con secuelas de hipoxia e hipoglucemia». Sugiere continuar manejo anticomicial y realización de nuevo ECG. Se realizan potenciales evocados auditivos de tallo cerebral, reportados como «normales bilaterales con mejor calidad de ondas del lado izquierdo, umbral auditivo de 30 dB bilateral». Reporte de potenciales evocados visuales: «anormales lado derecho con retardo de la vía visual de tipo desmielinizante y axonal, normales lado izquierdo». Se decide egreso del servicio el día 18 de abril con doble manejo anticomicial, cita a rehabilitación y seguimiento en consulta externa de pediatría.
DISCUSIÓN
Aroor et al (9) describieron tres casos de pacientes en edad pediátrica con crisis convulsivas que no remitieron con fármacos antiepilépticos de primera y segunda línea ni sedantes. El primer caso fue el de un niño de 7 años con fiebre y convulsiones persistentes manejado con DFH, fenobarbital, valproato, levetiracetam y clobazam; por persistencia de las crisis se procedió con infusión de midazolam y ventilación mecánica asistida. El análisis de líquido cefalorraquídeo (LCR) fue sugestivo de meningoencefalitis viral. El segundo caso fue el de una niña de 7 años con antecedentes de fiebre y crisis convulsivas tónico-clónicas; análisis de laboratorio y LCR fueron normales. Continuó presentando las crisis aún con manejo de DFH, fenobarbital, valproato, levetiracetam e infusión de midazolam y ketamina con ventilación mecánica asistida. El ECG continuó mostrando datos de actividad epileptogénica, por lo que se le añadió manejo empírico con piridoxina y sulfato de magnesio. Ante la falta de mejoría, se sospechó de encefalitis autoinmune y se manejó con metilprednisona y dieta cetogénica con respuesta inadecuada. El último caso fue el de una niña de 6 años con convulsiones generalizadas tónico-clónicas a pesar de manejo de DFH, fenobarbital, levetiracetam, lacosamida y clobazam. Análisis de sangre, LCR y resonancia magnética cerebral fueron normales. Se inició infusión con midazolam, y por sospecha de encefalitis autoinmune se inició metilprednisolona, inmunoglobulina intravenosa y dos ciclos de rituximab. Los pacientes presentaron secuelas neurológicas graves como persistencia de convulsiones focales y generalizadas, déficit cognitivo, cuadriparesia espástica, distonía y movimientos atetoides.
El EESR es un estadio patológico poco frecuente, considerado una emergencia médica (9,10). Debe de ser identificado a la brevedad para prevenir secuelas neurológicas en el paciente, así como para determinar tratamiento oportuno. De igual manera, se debe reconocer la etiología, las más comunes en pacientes pediátricos incluyendo las encefalitis autoinmunes o infecciosas, traumatismos craneoencefálicos, infecciones del SNC, isquemia, epilepsia y enfermedades metabólicas. En el caso descrito se tenía el antecedente de ingesta de fármacos hipoglucemiantes como causante de las crisis convulsivas, pero en el EER donde la etiología no puede ser establecida se deben considerar síndromes genéticos epilépticos. La monitorización continua del paciente mediante ECG es esencial para el diagnóstico y el manejo del EER/EESR (11).
En el EE participan alteraciones eléctricas del metabolismo, barrera hematoencefálica y funcionamiento neuronal que se presentan de forma sincrónica y pueden finalizar en muerte neuronal, debido a un desequilibrio entre una excesiva excitación neuronal y defectos en sistemas de inhibición de actividad neuronal (12). En el EER y particularmente en el EESR, los mecanismos responsables de la terminación de las convulsiones fracasan y se desarrollan procesos fisiopatológicos adicionales que conducen a la persistencia del EE (11).
A nivel celular, el EE intensifica la internalización de los receptores sinápticos de ácido γ-aminobutírico (GABA) tipo A, mientras que la función de los receptores sinápticos adicionales se mantiene. Este «tráfico de receptores» sináptico conduce a una reducción global de la actividad inhibitoria del GABA, desempeñando un papel clave en el desarrollo de la farmacorresistencia, y, por ende, la resistencia a benzodiacepinas (3,11). De igual forma, la persistencia de las convulsiones y el desarrollo de EESR pueden explicarse por la sensibilidad a la estimulación neuronal mediada por ácido N-metil-D-aspártico, la insuficiencia mitocondrial, el daño a la barrera hematoencefálica y la neuro inflamación (11). No existe un tiempo establecido para definir que una crisis epiléptica sea lesiva, estimándose entre 20 y 60 minutos (12).
En la actualidad no se cuenta con guías específicas para el manejo del EESR, basándose únicamente en la opinión experta o reporte de casos (5). Broomall et al (10) describieron dos casos de pacientes pediátricos en EESR manejados con allopregnalona, los cuales presentaron mejoría de las crisis tras su administración, pero requirieron continuar con manejo de tercera línea por ciertos periodos de tiempo. Algunos autores sugieren el uso de ketamina e hipotermia (13); otras sugerencias incluyen terapia inmunológica, dieta cetogénica, administración de piridoxina y neurocirugía (5,13). De igual manera se recomienda mantener la infusión de antiepilépticos de forma continua (5). Otras terapias emergentes incluyen la terapia electroconvulsiva y la estimulación magnética transcraneal (13); sin embargo, existe poca información sobre la eficacia y seguridad de estas terapias, por lo que sería recomendable ampliar los estudios sobre el manejo del EESR.
CONCLUSIONES
El EESR es una entidad clínica poco frecuente en pediatría, que debido a su alta mortalidad y morbilidad debe de ser diagnosticada de manera oportuna. No se cuenta con casos reportados de EESR secundario a hipoglicemia en pediátricos, tal como se reporta en este caso. Actualmente no hay descritos protocolos establecidos para el tratamiento del EESR; sin embargo, este no debe retrasarse debido a las probables complicaciones que el paciente puede presentar en caso de crisis convulsivas prolongadas.
Los autores no declaran conflicto de interés
Este estudio no recibió apoyo institucional
REFERENCIAS
1. Sequeira C, Chang J. Diagnóstico y Manejo de la Primera Convulsión, Rev Clin Esc Med. 2018; 8(2):11-21.
2. Cassel G, Beal J, Longani N, Leone B, Rivera R, Katyal C. Protocol-Driven Management of Convulsive Status Epilepticus at a Tertiary Children’s Hospital: A Quality Improvement Initiative, Pediatr Crit Care Med. 2019; 20(1):47–53.
3. Nelson S, Varelas P. Status Epilepticus, Refractory Status Epilepticus, and Super-refractory Status Epilepticus, Neurocrit Care. 2018; 24(6):1683-707.
4. Ferlisi M, Hocker S, Grade M, Trinka E, Shorvon S. Preliminary results of the global audit of treatment of refractory status epilepticus, Epilepsy Behav. 2015; 49:318-24.
5. Arayakarnkul P, Chomtho K. Treatment options in pediatric super-refractory status epilepticus, Brain Dev. 2018.
6. Meza O, Ochoa X. Fiesta neuronal: estado epiléptico en pediatría, An Med. 2018; 63(1):38-47.
7. Dhiren D, Krishna G. Supra-recommendation Treatment of Super-refractory Status Epilepticus, J Epilepsy Res. 2016; 6(1):39-41.
8. Lu W, Weng W, Wong L, Lee W. The etiology and prognosis of super-refractory convulsive status epilepticus in children. Epilepsy Behav. 2018; 86:66-71.
9. Aroor S, Shravan K, Mundkur S, Jayakrishnan C y Sripad S. Super-Refractory Status Epilepticus: A Therapeutic Challenge in Paediatrics, J Clin Diagn Res. 2017; 11(8), 1-4.
10. Broomall E, Natale J, Grimason M, Goldstein J, Smith C, Chang C, et al. Pediatric Super-Refractory Status Epilepticus Treated with Allopregnanolone, Ann Neurol. 2014; 76(6), 911-15.
11. Vasquez A, Farias R, Tatum W. Pediatric refractory, and super-refractory status epilepticus, Seizure: Eur J Epilepsy. 2018.
12. Castellanos R, Peralta E, Suárez J, Nariño D. Enfoque del estatus epiléptico en adultos: consideraciones sobre la fisiopatología y tratamiento, Acta Neurol Colomb. 2017; 33(3): 199-210.
13. Marawar R, Basha M, Mahulikar A, Desai A, Suchdev, Shah A. Updates in Refractory Status Epilepticus, Crit Care Res Pract. 2018. 1-19.



