
FIRST CASE OF CRI DU CHAT SYNDROME IN THE STATE OF QUINTANA ROO
Marleene Eunice Pedroza Guardado1, Alejandro Gaviño Vergara2
1 Estudiante terapia física
2 Centro de Rehabilitación Infantil Teletón (CRIT). Quintana Roo. México.
Correspondencia: Alejandro Gaviño-Vergara. Correo contacto: Esta dirección de correo electrónico está protegida contra spambots. Usted necesita tener Javascript activado para poder verla.
Recibido: 08 de enero de2019.
Aceptado: 22 de mayo de 2019.
RESUMEN
Introducción. El síndrome de Cri Du Chat es una anomalía genética rara causada por la pérdida de material genético en el brazo corto del cromosoma 5. Tiene una incidencia de 1 por cada 50,000 nacidos vivos. Sus principales manifestaciones clínicas son microcefalia, llanto agudo, retraso mental, hipotonía, cara redonda, entre otras. Este es el primer caso reportado en el estado de Quintana Roo.
Caso clínico. Paciente femenino de cinco años de edad, control prenatal regular, peso al nacer de 2.9 kg, talla 48 cm, APGAR de 9/10 diagnosticada por médico genetista a los tres años de edad por estudio citogenético de cariotipo en sangre periférica.
Conclusión. La importancia de conocer las principales características de este síndrome tan poco frecuente, nos lleva al diagnóstico oportuno de los pacientes, los cuales deben recibir una rehabilitación integral para lograr desarrollar al máximo sus capacidades. Esto mejorará la independencia del paciente y la integración en la sociedad.
Palabras clave: Monosomía; Cromosomas Humanos Par 5; Deleción Cromosómica
ABSTRACT
Introduction. Cri Du Chat syndrome is a rare genetic abnormality caused by the loss of genetic material in the short arm of chromosome 5. It has an incidence of 1 per 50,000 live births. Its main clinical manifestations are microcephaly, acute crying, mental retardation, hypotonia, round face, among others. This is the first case reported in the state of Quintana Roo.
Clinical case. Female patient of five years of age, regular prenatal control, birth weight of 2.9 kg, height 48 cm, APGAR of 9/10 diagnosed by medical geneticist at three years of age by cytogenetic study of karyotype in peripheral blood.
Conclusion. The importance of knowing the main characteristics of this infrequent syndrome leads us to the timely diagnosis of patients, they must receive a comprehensive rehabilitation to achieve maximum development of their capabilities. This improves patient independence and integration into society.
Key words: Monosomy; Chromosomes, Human, Pair 5; Chromosome Deletion
INTRODUCCIÓN
El síndrome del deleción 5p o maullido de gato (del francés Cri du Chat), o síndrome de Lejeune, es un trastorno genético raro que fue descubierto y clasificado en 1963 por el médico francés Lejeune y que es causado por la deleción (pérdida de un fragmento) del brazo corto de uno de los cromosomas 5 (5p-) (1). Se caracteriza por múltiples anomalías congénitas como: bajo peso al nacimiento, retraso mental, microcefalia, hipotonía, facies características y un llanto muy característico similar al de un maullido de gato; el fenotipo es cambiante y evolutivo según la edad de los pacientes (2,3).
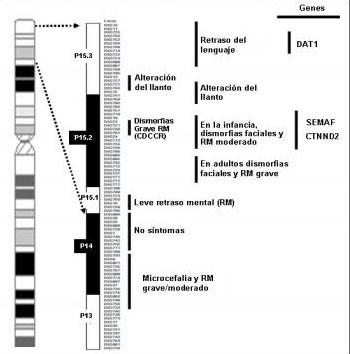
La incidencia de esta entidad clínica varía entre 1 por cada 15,000 y 1 por cada 50,000 recién nacidos vivos, y en la población con retraso mental puede llegar a ser hasta de 1 por cada 3,500 nacidos vivos (4), lo que constituye uno de los síndromes más comunes por deleción del grupo B cromosómico 5. La mayoría de los casos se debe a deleciones de novo y, menos frecuente, a translocaciones balanceadas parentales o aberraciones cromosómicas raras (3,6). La pérdida de material genético puede variar desde fragmentos tan pequeños como la región 5p15.2 (región crítica del síndrome) hasta el brazo corto completo del cromosoma 5. Se ha comprobado una correlación entre las características fenotípicas y el tamaño de la deleción (7,8) (figura 1).
Figura 1. Región crítica. Localización de ciertos defectos
CASO CLINICO
Paciente femenino de cinco años ocho meses de edad, producto del sexto embarazo, antecedentes de dos abortos previos, madre de 38 años y padre de 47 años al nacimiento, aparentemente sanos, no consanguíneos, originarios de Valladolid, Yucatán.
Es obtenida a las cuarenta semanas de gestación por parto via vaginal con peso al nacer de 2.9 kg, talla 48 cm, APGAR de 9/10 y respiración espontanea, la cual es egresada como sana, dada de alta a las veinticuatro horas.
A los dos meses de edad se le realizó cirugía de hernio plastia umbilical, posterior a eso estuvo hospitalizada 3 semanas por cuadro de neumonía. A los tres meses de edad llama la atención a los padres bajo tono muscular en ambas extremidades así como llanto agudo y débil. Acuden al médico particular manejada con diagnóstico de retraso global del neurodesarrollo con hipotonía, por lo cual acudió a recibir servicio de terapia física y ocupacional al Centro de Rehabilitación Infantil Municipal (CRIM) de Cancún a partir de los primeros meses de edad.
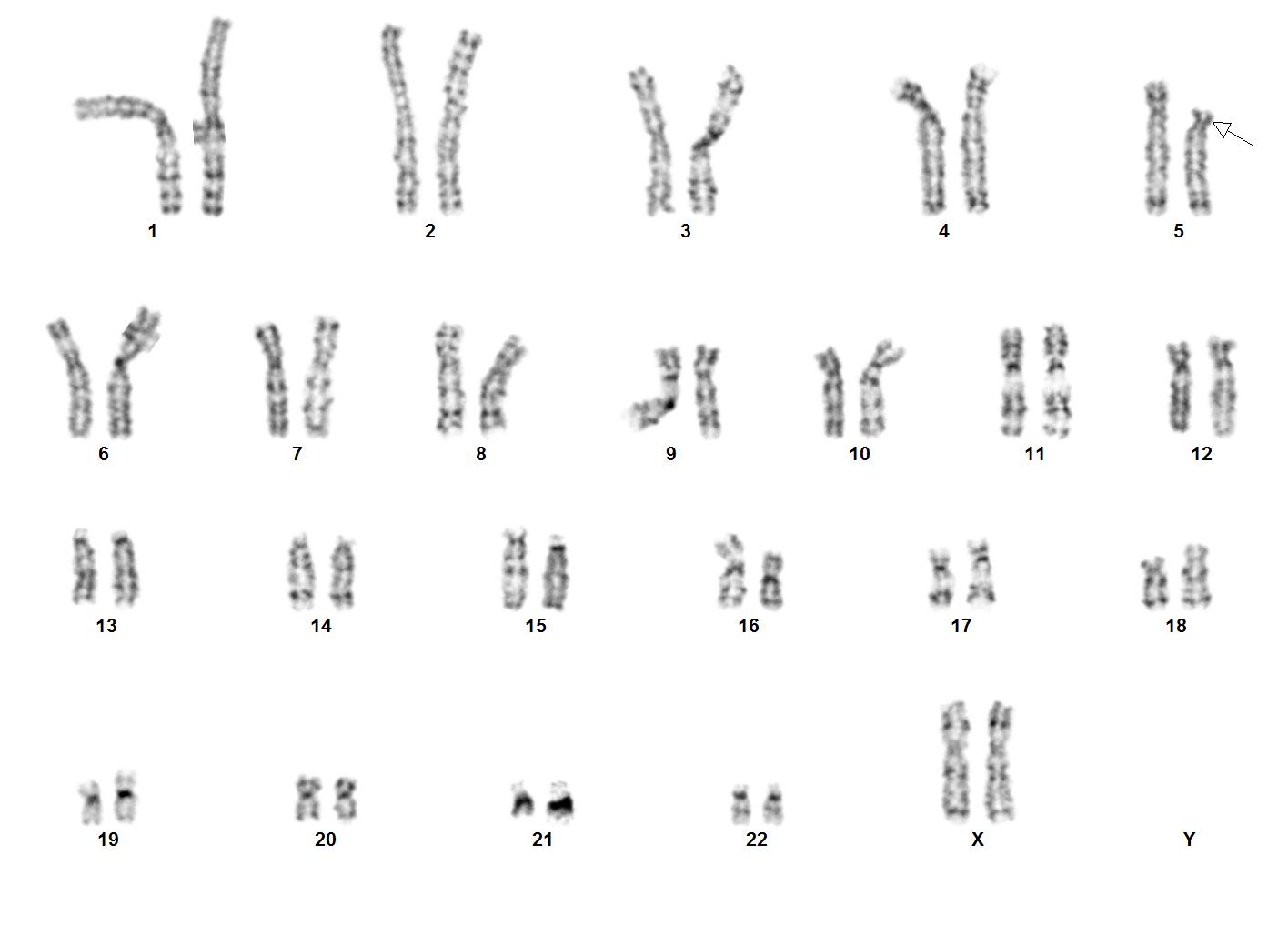
A los dos años siete meses ingresa al Centro de Rehabilitación Infantil Teletón (CRIT) de Cancún, en donde al ser evaluada por el médico genetista y revisión de dismorfias como microcefalia, cara redonda, epicanto, antecedente de llanto agudo, solicita cariotipo en sangre periférica bandas G con atención en región 5p , confirmando el diagnóstico de esta entidad a los tres años de edad. Fórmula resultado 46,XX, del (5)(p15.2) (figura 2) .
Figura 2. Deleción cromosoma 5 brazo corto, marcado con la flecha 46,XX, del (5)(p15.2)
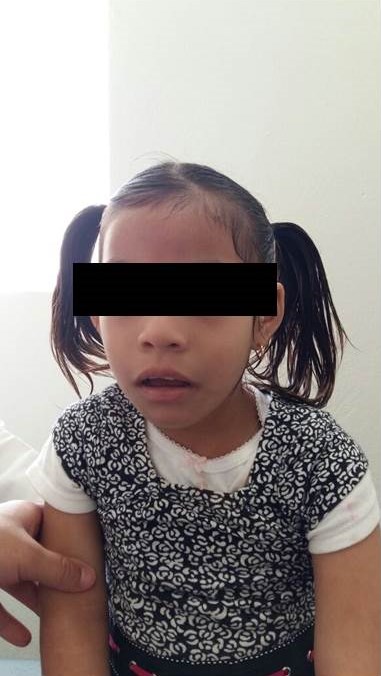
Exploración física 2 años 7 meses Peso: 10.6Kg. (p <3) Talla: 86cm .(p 3-10) Perímetro cefálico (P. C.): 43cm.(p<3) Circunferencia de brazo (C. B.): 0cm. Cráneo con microcefalia, cara redonda, fisuras palpebrales ligeramente oblicuas hacia arriba, epicanto bilateral, puente nasal ancho, retrognatia, paladar alto y ojival, pabellones auriculares con apéndice preauricular izquierdo, tórax con ruidos cardiacos sin alteraciones, abdomen con cicatriz quirúrgica , no se palpan hernias ni megalias, genitales externos femeninos, hipotonía generalizada, miembros superiores con presencia de pliegue transverso en palma mano izquierda, miembros inferiores sin otras alteraciones, manchas azuladas en dorso así como hipertricosis.
Figura 3. Dismorfias faciales síndrome de Cri du Chat
Desarrollo psicomotor. Sostén cefálico 12 meses, sedestación 14 meses, gateo 2 años, bipedestación 2 años 2 meses, marcha 3 años 6 meses, pinza gruesa de 18 meses, pinza fina 20 meses, balbuceo no recuerda, sonrisa social 12 meses, ingiere alimentos de manera independiente a los 4 años, pronuncia bisilábicos sin lograr oraciones, identifica números y colores de manera visual.
Actualmente la paciente tiene 5 años 9 meses de edad, presenta adecuado seguimiento visual y auditivo, realiza marcha independiente con alteraciones en las fases de la marcha, ascenso y descenso de escaleras con alternancia. Presenta genu recurvatum de ambas rodillas, equilibrio deficiente, mala alineación postural global. En la parte de lenguaje logra bisilábicos, ingiere alimentos de manera independiente, tiende a la autoagresión de manera frecuente, identifica miembros de su familia y personas cercanas a su entorno, presenta problemas de conducta. Se encuentra integrada a un CAM (centro de atención múltiple) desde que tenía 1 año y 6 meses de edad.
DISCUSIÓN
El presente reporte es el primer caso de síndrome de Cri Du Chat confirmado en el estado de Quintana Roo, por lo que los autores consideran importante realizar la descripción del caso clínico y así conocer la información que se encuentra actualmente sobre esta entidad. El diagnóstico de la paciente fue hecho a los 3 años de edad, debido a que las dismorfias faciales y su llanto fueron las características principales que llamaron la atención del médico especialista en genética.
La paciente hasta el momento ha tenido un manejo integral en donde ha recibido atención de especialistas de área médica en el CRIT Quintana Roo, terapia física, terapia ocupacional, terapia pulmonar, terapia de lenguaje, terapia psicológica, nutrición y psicopedagogía. El diagnóstico temprano y la rehabilitación integral son fundamentales para mejorar la calidad de vida de los pacientes con Cri Du Chat (15). Aunque en la actualidad no existe un tratamiento específico para este síndrome, es importante que los pacientes reciban un manejo integral, debido a que existe retraso global en el neurodesarrollo, lo que puede mejorarse para aumentar la independencia del paciente.
Para los niños que presentan el clásico llanto similar al maullido de un gato o con retraso mental y anomalías congénitas, el diagnóstico se debe confirmar lo antes posible realizando un estudio citogenético o cariotipo que se realiza mediante la técnica para bandas G convencional. Si los resultados del estudio citogenético son de apariencia normal o no corresponden con la clínica del paciente ante la sospecha, es necesario hacer un análisis citogenético molecular más específico denominado fluorescencia por hibridación in situ (FISH) (14). Los análisis moleculares han demostrado que el cromosoma que presenta la deleción, en la mayoría de los casos, es el de origen paterno (11,12,13). Hay controversia sobre si el tamaño de la deleción se asocia con la gravedad del retraso mental (16), ya que-, según algunas observaciones, éste era desproporcionadamente grave respecto al tamaño de la deleción (17).
A continuación se comparan las manifestaciones clínicas del paciente con el cuadro clásico descrito de síndrome de 5p-.
|
Anormalidades |
Porcentaje |
Paciente |
|
Bajo peso al nacer (menor a 2.5kg.) |
72% |
|
|
Crecimiento lento |
100% |
* |
|
Llanto en forma de gato |
100% |
* |
|
Retraso mental |
100% |
* |
|
Hipotonia |
78% |
* |
|
Microcefalia |
100% |
* |
|
Cara redonda |
68% |
* |
|
Hipertelorismo |
94% |
|
|
Pliegues epicanto |
85% |
* |
|
Fisuras palpebrales oblicuas hacia abajo |
81% |
|
|
Estrabismo, a menudo divergente |
61% |
|
|
Baja implantación pabellones auriculares |
|
|
|
Asimetrías faciales |
-- |
* |
|
Enfermedades congénitas cardiacas |
30% |
|
|
Pliegue simiano |
81% |
. |
|
Falange triradial |
40% |
|
|
Acortamiento metacarpianos |
-- |
|
| Características de la paciente en el 1 er año de vida: | Características de la paciente en la actualidad: | ||
|
|
La tasa de supervivencia y la esperanza de vida son altas, teniendo registro de casos de individuos que superan los 50 años de edad. La morbimortalidad es mayor durante los primeros años: el 75% de las muertes registradas ocurrieron en los primeros meses tras el nacimiento, y un 90% tuvo lugar en el primer año de vida (15,19- 24). El punto más delicado y que preocupa más a las familias es la alta posibilidad de que los niños con Cri du Chat sean incapaces de valerse por sí solos y desenvolverse socialmente. Sólo la mitad de los niños adquieren las habilidades verbales suficientes para comunicarse. En la parte física, otras preocupaciones están relacionadas con las habituales complicaciones respiratorias y óticas que precisan incluso de ingreso hospitalario (23).
No se encuentran datos del número de casos diagnosticados en México, se estima que cada año nacen 20 niños con este síndrome en nuestro país, se hace una invitación a informar sobre el desarrollo de los casos.
CONCLUSIÓN
Este trabajo presenta los principales datos clínicos para sospecha de este síndrome de Cri Du Chat para un diagnóstico temprano y oportuno, para que el paciente tenga un tratamiento adecuado y eficaz.
Se recomienda a los padres que estén interesados en un nuevo embarazo, realizarse el estudio de FISH, para detectar o confirmar anomalías génicas o cromosómicas que generalmente están más allá de la capacidad de resolución de la citogenética de rutina.
Gracias a las recientes mejoras en el manejo de los pacientes con síndrome de Cri du Chat (como la aplicación de los programas de rehabilitación), orientaciones generales para la asistencia han permitido mejorar el desarrollo psicomotor, la autonomía y la adaptación social de estos pacientes para así favorecer su integración en la sociedad actual.
REFERENCIAS
1. Nardi, S. (2014). El síndrome de maullido de gato, aspectos característicos y pautas educativas. Disponible en : http://www.criduchat.it/documents/El-sindrome-del-maullido-de-gato-Aspectos-caracteristicos-web.pdf .
2. Grouchy de J, Turleau C. Atlas des malades chromomiques, 10.ª ed. París: Expansion Scientifique Française, 1982; 54-57.
3. Niebuhr E. The cri du chat syndrome: epidemiology, cytogenetics, and clinical features. Hum Genet. 1978; 44: 227-275.
4. Harvard C, Malenfant P, Koochek M, Creighton S, Mickelson EC, Holden JJ, et al. A variant cri du chat phenotype and autism spectrum disorder in a subject with de novo cryptic microdeletions involving 5p15.2 and 3p24.3-25 detected using whole genomic array CGH. Clin Genet. 2005; 67(4): 341-351.
5. Correa-Nazco VJ, Romero-Pérez JC, Martín V, Laynez-Cerdeña P, Minguélez-Morales M, Linares-Feria L. Síndrome de Wolf-Hirschhorn. Rev Neurol. 1999; 28(9): 925-930.
6. Romano C, Ragusa RM, Scillato F, Greco D, Amato G, Barcelleta C. Phenotypic and phoniatric findings in mosaic cri du chat syndrome. Am J Med Genet. 1991; 39: 391-395.
7. Overhauser J, Huang X, Gersh M, et al. Molecular and phenotypic mapping of the short arm of chromosome 5: sublocalization of the critical region for the cri du chat syndrome. Human Mol Genet. 1994; 3: 347-352.
8. Cerruti Mainardi P, Perfumo P, Cali A, et al. Clinical and molecular characterization of 80 patients with 5p deletion: genotype-phenotype correlation. J Med Genet. 2001; 38: 151-158.
9. Nardi, S. (2014). El síndrome de maullido de gato, aspectos característicos y pautas educativas. Recuperado de: http://www.criduchat.it/documents/El-sindrome-del-maullido-de-gato-Aspectos-caracteristicos-web.pdf.
10. M. L. Martínez-Fernández, D. Sánchez Izquierdo, M. L. Martínez-Frías. Síndrome de Deleción 5p. Julio 2010. Disponible en: https://www.orpha.net/data/patho/Pro/es/SindromeCriDuChat.pdf
11.Zarina A L, Juriza L, Sharifah A Z, Azli L, Chia W K, Khairunisa K, et al. Cri du chat Syndrome: Application of Array CGH in Diagnostic Evaluation. Med & Health 2010;5(2):108-13.
12. Azman B Z, Akhir S M, Zilfalil B A, Ankathil R. Two cases of deletion 5p syndrome: one with paternal involvement and another with atypical presentation. Singapore Med J. 2008;49(4):e9.
13. Torun D, Bahce M, Alanbay I, Guran S, Baser I. Prenatal diagnosis of Cri du chat syndrome following high maternal serum human chorio
14. Blanca L. Galo, Ramón H. Alvarenga, Sindrome de Cri du chat una rara cromosomopatía, REV MED HONDUR, Vol. 80, No. 1, 2012, pag 17-19
15. Caro,N. (2013). Estudio psicopedagógico del Síndrome del Maullido del Gato ., de asimaga Sitio web: https://asimaga.org/estudio-psicopedagogico-del-sindrome-del-maullido-del-gato/.
16. Zhang X, Snijders A, Segraves R, et al. High-resolution mapping of genotype-phenotype relat cri du chat syndrome using array comparative genomic hybridization. Am J Hum Genet. 2005; 76: 312-326.
17 Marinescu RC, Johnson EL, Grady D, Chen X, Overhauser J. FISH analysis of terminal deletions in patients diagnosed with cri-duchat syndrome. Clin Genet. 1999; 56: 282-288.
18. Gómez, E., Olivar, G.,García, E., Domínguez, M. & Arias, O.. (2011). Manejo inicial del Síndrome de Cri du Chat. 01 Sep 2018, de Vox Paediatrica Sitio web: https://spaoyex.es/sites/default/files/pdf/Voxpaed18.1pags97-100.pdf
19. Cerruti Mainardi P, Medalogo LM, Pedrianazzi Mº. Cri du Chat síndrome. Orphanet J Rare Dis. 2006 Sep5; 1: 33.
20. Mainardi PC, Perfumo C, Calì A, Coucourde G, Pastore G, Cavani S. Clinical and molecular characterisation of 80 patients with 5p deletion: genotype-phenotype correlation. J Med Genet. 2001;38:151-8.




