X-LINKED HYDROCEPHALUS. REPORT OF TWO CASES
Alejandro Gaviño-Vergara1, Alma Rubí Lemarroy-Toledo2, Lilian Citlali Lara-Solano2
1 Centro de Rehabilitación e Inclusión Infantil Quintana Roo. México.
2 Estudiante de pregrado de Medicina. Universidad Anáhuac Cancún. México.
Correspondencia: Alejandro Gaviño-Vergara. Centro de Rehabilitación e Inclusión Infantil Quintana Roo. Correo electrónico: drgavino@live.com.mx
Recibido: 15 de febrero de 2017.
Aceptado: 10 de abril de 2017.
RESUMEN
Introducción. La hidrocefalia ligada al X es un desorden genético localizado en el cromosoma X, el cual se caracteriza por las siguientes manifestaciones clínicas: retraso mental, pulgares aductos bilaterales y espasticidad en miembros superiores e inferiores. Es la forma genética más común de hidrocefalia congénita, con una incidencia de 1 por cada 30.000 nacimientos en el caso de varones.
Caso clínico. Se presentan los casos clínicos de dos hermanos varones de 15 y 16 años de edad con el diagnóstico de hidrocefalia ligada al X. Tienen antecedentes familiares de un tío, de rama materna, fallecido con el mismo diagnóstico. Se dio asesoramiento genético a la familia debido a la herencia recesiva ligada al X y se dio seguimiento a posibles mujeres portadoras.
Discusión. La importancia de este caso clínico radica en el seguimiento y asesoramiento genético a los familiares, ya que el árbol genealógico de los pacientes muestra un claro patrón de herencia recesivo ligado al X. Es básico el tratamiento y seguimiento en estos pacientes por un equipo multidisciplinario, con experiencia en diferentes áreas como: pediatría, neurología, neurocirugía, rehabilitación y genética.
Conclusión. La hidrocefalia ligada al X es una entidad que deben conocer tanto médicos generales como especialistas, ya que además de repercutir en el seguimiento y diagnóstico de estos pacientes se debe estudiar a posibles portadoras de la enfermedad dentro de la misma familia.
Palabras clave: hidrocefalia, cromosoma X, asesoramiento genético
ABSTRACT
Introduction. The X-linked hydrocephalus is a genetic disorder located on the X chromosome, which is characterized by the following clinical manifestations: mental retardation, bilateral adducted thumbs and spasticity in the upper and lower limbs. It is the most common genetic form of congenital hydrocephalus, with an incidence of 1 per 30.000 births in the case of males.
Clinical case. We present the case of two brothers of 15 and 16 years of age with the diagnosis of X-linked hydrocephalus. They have a family history of an uncle, of maternal branch, who died with the same diagnosis. Genetic counseling was given to the family due to the X-linked recessive inheritance and followed possible women carriers.
Discussion. The importance of this clinical case lies in the follow-up and genetic counseling to the relatives, since the genealogical tree of the patients shows a clear pattern of X-linked recessive inheritance. It is basic the treatment and follow-up in these patients by a multidisciplinary team, with experience in different areas such as: pediatrics, neurology, neurosurgery, rehabilitation and genetics.
Conclusion. X-linked hydrocephalus is an entity that must know both general practitioners and specialists, since in addition to having an impact on the follow-up and diagnosis of these patients should be studied to possible carriers of the disease within the same family.
Key words: hydrocephalus, X chromosome, genetic counseling
INTRODUCCIÓN
La hidrocefalia es la acumulación excesiva de líquido cefalorraquídeo (LCR) generada por un desorden hidrodinámico; ya sea en la producción, circulación o reabsorción de LCR, dando como resultado la dilatación de los ventrículos (1). Su clasificación desde el punto de vista etiológico se divide en dos: congénita, la cual está presente al nacimiento y casi siempre asociada con problemas en el desarrollo; y adquirida, la cual ocurre después de la formación del cerebro y los ventrículos (2).
Dentro de las causas congénitas, la hidrocefalia ligada al cromosoma X es la causa genética más común, con una incidencia de 1 por cada 30.000 nacimientos en varones, y se define como un desorden genético, el cual se caracteriza por las siguientes manifestaciones clínicas: retraso mental, pulgares aductos bilaterales, así como espasticidad en miembros superiores e inferiores (3,4).
Dentro de los diagnósticos diferenciales de esta patología hay formas síndromáticas y no sindromáticas (5). Un ejemplo de la forma sindromática puede incluir alteraciones cromosómicos o algunas condiciones con un patrón de herencia mendeliano, en el cual ya existe un gen identificado. Por otro lado, está la hidrocefalia congénita no sindromática, la cual puede estar asociada a un defecto del tubo neural, malformación del sistema nervioso central o secundaria a una hemorragia, entre otras.
A continuación se presenta el reporte de caso de dos hermanos con antecedentes familiares de un tío de rama materna, con el diagnóstico de hidrocefalia ligada al cromosoma X.
CASOS CLÍNICOS
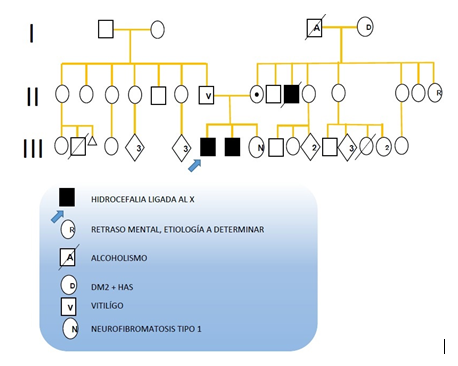
Pacientes masculinos de 15 y 16 años de edad. Cuentan con los siguientes antecedentes familiares de importancia (figura 1):
Figura 1. Árbol genealógico de pacientes, en el cual se puede observar el antecedente de hermano con mismo diagnóstico del caso índice, así como tío rama materna fallecido por hidrocefalia ligada al X. Por otro lado hay un antecedente de neurofibromatosis tipo 1 de una de las hermanas.
Caso 1.
Masculino de 15 años de edad que cuenta con los siguientes antecedentes: madre y padre originarios de Yucatán, sin antecedentes familiares de consanguinidad o endogamia. Producto de la gesta II/III, embarazo con control prenatal regular a partir del tercer mes de embarazo. Por ultrasonido realizado en séptimo mes se reporta hidrocefalia y circular de cordón a cuello, la madre niega toxicomanías y complicaciones durante el embarazo. El producto es obtenido por cesárea de término con peso de 3.2 kg, talla 50 cm, APGAR 8/9. A los 15 días de vida requirió colocación de válvula de derivación ventriculoperitoneal.
El desarrollo psicomotor logra sostén cefálico al año de edad, sedestación 4 años, no logra bipedestación. El lenguaje es a base de bisílabos. Actualmente el paciente presenta epilepsia clónica generalizada en manejo con valproato de magnesio. Cuenta con estudio de tomografía axial computarizada cerebral que reporta hidrocefalia y dilatación de ventrículos laterales con colocación de válvula ventrículo peritoneal.
La exploración física se presenta en las figuras 2, 3 y 4.
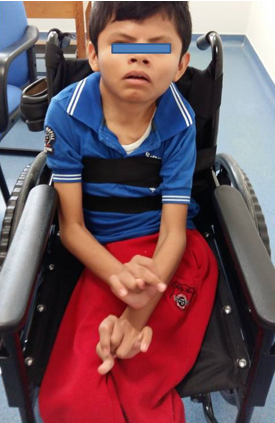
Figura 2. Masculino 15 años de edad con cráneo postoperado válvula derivación ventriculoperitoneal , arcos supraciliares prominentes, cejas escasas, puente nasal ancho, comisuras labiales hacia abajo, espasticidad en miembros superiores con pulgares en aducción.
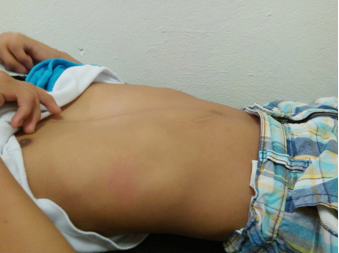
Figura 3. Tórax con pectum excavatum.
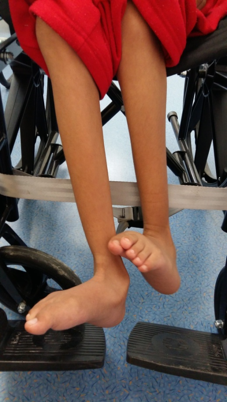
Figura 4. Espasticidad en miembros inferiores.
Caso 2.
Paciente masculino de 16 años de edad producto de la gesta I/III, la gesta II corresponde a hermano de 15 años con diagnóstico de hidrocefalia ligada al X. La madre presentó embarazo con control prenatal regular a partir del mes de edad, se realizó ultrasonido al quinto mes el cual reportó hidrocefalia. La madre niega antecedentes toxicológicos o complicaciones durante el embarazo. Producto obtenido de término por cesárea debido a detección de hidrocefalia de manera prenatal con peso de 3.0 kg, talla 50 cm, APGAR:8/9 con llanto y respiración espontanea.
Al nacimiento se confirma hidrocefalia y a los 20 días de nacimiento fue operado con válvula de derivación ventriculoperitoneal. Presentó desarrollo psicomotor con retraso global en el neurodesarrollo; el lenguaje a base de balbuceo y algunos monosílabos. La exploración física actual se presenta en las figuras 5 y 6.
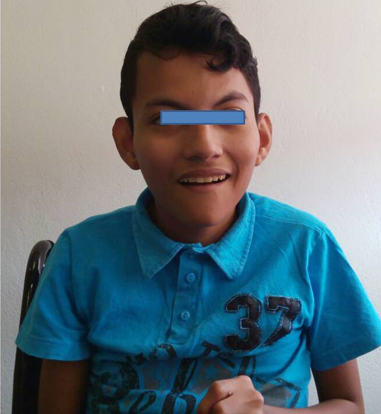
Figura 5. Cráneo postoperado con colocación de válvula ventrículo-peritoneal, cara alargada, frente alta, fisuras palpebrales oblicuas hacia arriba, puente nasal ancho con pabellones auriculares acopados.
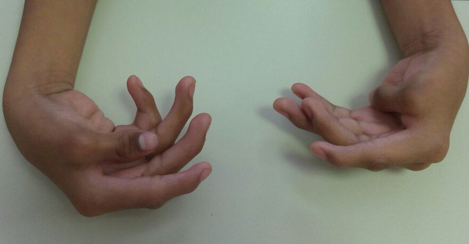
Figura 6. Contracturas en ambas manos con ambos pulgares en aducción.
DISCUSIÓN
Dentro de los casos de hidrocefalia congénita asociado a un patrón de herencia característico, el mecanismo de herencia recesivo ligado al cromosoma X es el más frecuentemente reportado dentro de las formas hereditarias en el paciente con hidrocefalia. En la mayoría de los casos, mutaciones en el gen L1CAM generan un defecto de la actividad en las moléculas de adhesión celular neuronal, lo cual se correlaciona con la afección neurológica en estos pacientes. El L1CAM se encuentra principalmente en el sistema nervioso de algunas especies entre ellas el ser humano (6,7).
La importancia de este caso clínico radica en el seguimiento y asesoramiento genético a la familia, ya que el árbol genealógico muestra un patrón de herencia recesivo ligado al cromosoma X, en el cual las mujeres portadoras de esta entidad tienen la probabilidad de un 50% de transmitir la enfermedad a su descendencia. En el caso de varones tienen un 50% de riesgo de estar sanos o enfermos. En este caso la madre de ambos pacientes es una portadora obligada al tener 2 hijos afectados y un hermano fallecido con el mismo cuadro clínico. Debido a esto se otorgó el asesoramiento genético a los familiares. Se aconsejó que dentro de las opciones será importante realizar estudio molecular a todas las hermanas de la madre para conocer si son portadoras y conocer los riesgos de recurrencia ya comentados en caso de serlo.
En cuanto al manejo médico es básico el tratamiento y seguimiento en estos pacientes por un equipo multidisciplinario, con experiencia en diferentes áreas como: pediatría, neurología, neurocirugía, rehabilitación y genética. Los pacientes de este caso tienen el seguimiento de manera periódica. La colocación de una válvula de derivación de líquido cefalorraquídeo puede realizarse en caso de ser necesario para reducir la presión intracraneal, procedimiento que se realizó en edades tempranas con ambos hermanos. La cirugía para pulgares aductos también debe ser valorada por el especialista en ortopedia, ya que muchas veces no está justificado realizarlo, esto dependerá de la funcionalidad de cada caso. En cuanto a la discapacidad intelectual el progreso del desarrollo deber ser monitoreado y vigilado estrechamente. Finalmente para prevenir complicaciones secundarias es conveniente llevar a cabo rehabilitación física, la evaluación y seguimiento neurológico con el objetivo de mejorar tono muscular y espasticidad (8).
CONCLUSIÓN
La hidrocefalia ligada al cromosoma X es una entidad que deben conocer tanto médicos generales como especialistas, ya que además de requerir un seguimiento y valoración multidisciplinaria, el conocer la historia y antecedentes familiares puede apoyar en el asesoramiento genético en cada uno de los miembros de la familia para conocer los riesgos de recurrencia en caso de nuevos embarazos.
REFERENCIAS
1. Sevillano-García MD, Cacabelos-Pérez P, Cacho-Gutiérrez J. Alteraciones del líquido cefalorraquídeo y de su circulación: hidrocefalia, pseudotumor cerebral, síndrome de presión baja. Medicine 2011; 10(71): 4814-24.
2. Zhang J, Williams M, Rigamonti D. Genetics of human hydrocephalus. J Neurol 2006;253:1255–1266.
3. Ferese R, Zampatti S, Griguoli AMP, Fornai F, Giardina E, Barrano G, et al. A New Splicing Mutation in the L1CAM Gene Responsible for X-Linked Hydrocephalus (HSAS). Journal of Molecular Neuroscience 2016; 1-6. Disponible en: http://dx.doi.org/10.1007/s12031-016-0754-3.
4. Basel-Vanagaite L, Straussberg R, Friez MJ, Inbar D, Korenreich L, Shohat M, et al. Expanding the phenotypic spectrum of L1CAM-associated disease. Clin Genet 2006;69:414–9.
5. Verhagen JMA. et al. Congenital hydrocephalus in clinical practice: a genetic diagnostic approach. Eur J Med Genet. 2011 Nov-Dec;54(6):e542-7. Disponible en: http://dx.doi.org/10.1016/j.ejmg.2011.06.005.
6. Kenwrick S, Watkins Al, De Angelis E. Neural cell recognition molecule L1: relating biological complexity to human disease mutations. Hum Mol Genet 2000; 9 (6): 879-886. Disponible en: http://dx.doi.org/10.1093/hmg/9.6.879
7. De Angelis E, Watkins A, Schafer M, Brummendorf T, Kenwrick S. Disease-associated mutations in L1 CAM interfere with ligand interactions and cell-surface expression. Hum Mol Genet. 2002;11:1–12.
8. Pagon RA, Adam MP, Ardinger HH, et al. editors. GeneReviews Seattle (WA): University of Washington, Seattle; 1993-2017. [citado febrero 2017] Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1484.
