
MORQUIO´S SYNDROME: CASE REPORT
Oscar Rosaldo-Abundis1, David Alejandro Pasos-Aldaz2, Zaazil Pilar Jiménez-Maldonado2
1 Hospital General de Chetumal. Secretaría de Salud de Quintana Roo. México.
2 Estudiante de la División de Ciencias de la Salud. Universidad de Quintana Roo. México.
Correspondencia: Zaazil Pilar Jiménez-Maldonado. Retorno Carranza No. 531 “A”. Colonia Flamboyanes, Chetumal, Quintana Roo. México C.P. 77034. Correo electrónico: zaaziljm26@hotmail.com
Recibido: 01 de abril de 2016.
Aceptado: 08 de julio de 2016.
RESUMEN
Introducción. El síndrome de Morquio forma parte de un grupo raro de enfermedades de depósito lisosomal, sus características clínicas son resultado del déficit de la N- acetil- galactosamina-6- sulfatasa.
Caso clínico. Se presenta el caso de un hombre de 35 años de edad con fiebre, tos con expectoración, disnea y cianosis peribucal. Veinticuatro horas posteriores a la sintomatología inicial se agrega dolor abdominal localizado en cuadrante inferior derecho.
Discusión. Las manifestaciones clínicas y comorbilidades asociadas al síndrome de Morquio son secundarias a los depósitos de glicosaminoglicanos presentes en las células. Su incidencia es rara, y a pesar de haber casos que reportan diferentes patologías secundarias a la acumulación de estas sustancias, aún falta información de éste síndrome.
Palabras clave: mucopolisacaridosis IV, neumonía, hipotiroidismo
ABSTRACT
Introduction. Morquio's syndrome forms part of a rare group of lysosomal storage diseases, its clinical characteristics are a result of a galactosamine (N-acetyl)-6-sulfatase deficit.
Clinical Case. We present the case of a 35-year-old man with fever, cough with expectoration, difficulty to breathe and cyanosis peribucal. Twenty-four hours later to the initial symptomatology an abdominal pain located in low right quadrant is added.
Discussion. The clinical manifestations and comorbidities associated with Morquio's syndrome are secondary to the storage of glicosaminoglicanos present in the cells. Its incident is rare and in spite of the cases reporting different pathologies secondary to the accumulation of these substances, still lacking information of this syndrome.
Key words: mucopolysaccharidosis IV, pneumonia, hypothyroidism.
INTRODUCCIÓN
Las mucopolisacaridosis (MPS) son trastornos de almacenamiento lisosomal, causados por la deficiencia de las enzimas necesarias para la descomposición progresiva de glicosaminoglicanos (GAGS), lo que resulta en la disfunción celular y anormalidades clínicas. Este grupo de enfermedades raras tiene una incidencia total estimada -incluyendo todos los tipos de MPS- de aproximadamente 1 de cada 20.000 nacidos vivos.
Entre los años 1900 y 1913, John Thompson realizó la primera descripción de un caso de MPS, y en 1917, Charles Hunter publicó por primera vez sobre este grupo de enfermedades (1,2).
La MPS tipo IV, también conocida como síndrome de Morquio, es una enfermedad autosómica recesiva que consiste en dos formas (A y B) con resultados clínicos similares. Su etiología es resultado de mutaciones en el gen que codifica la galactosamina-6-sulfatasa (GALNS), o por deficiencia de beta-galactosidas (3). La MPS tipo IV fue descrita en 1929 por Morquio en Montevideo, Uruguay. Sin embargo, Brailsford en Inglaterra, caracterizaba de forma simultánea esta patología, por lo que este cuadro se denomina síndrome de Morquio-Brailsford (4).
El síndrome de Morquio es una de las MPS más frecuentes en México. Se estima que éste síndrome ocurre en 1 por cada 75.000 a 1 por cada 200.000 nacimientos, afecta tanto a hombres como a mujeres por igual y no hay predilección de raza (5).
Las características clínicas de este padecimiento son el resultado de la acumulación de sulfato de queratano y la condroitina-6-sulfato, y son de aparición progresiva, principalmente entre el primer a tercer año de vida. Las principales características del síndrome son las anormalidades esqueléticas: baja talla (particularmente de tronco corto), tórax en tonel y pectus carinatum, genu valgum e hiperlaxitud articular. También pueden ocurrir opacidades corneales leves, hepato-esplenomegalia y enfermedad cardíaca valvular. El crecimiento está muy comprometido antes de los 5 años de edad y la estatura promedio se encuentra entre los 85 y los 100 cm, pero no se presenta retardo mental como en las demás mucopolisacaridosis (6,7,8).
La prevención de las principales complicaciones mejora la calidad y expectativa de vida de los afectados. En la forma grave de la enfermedad, el crecimiento y la talla es mínima después de los seis o siete años de edad, y la muerte ocurre generalmente en la tercera o cuarta década por insuficiencia cardiorrespiratoria (11). La expectativa de vida general se encuentra entre la tercera y cuarta décadas de la vida; sin embargo, se han reportado casos de supervivencia hasta los 70 años (12).
DESCRIPCIÓN DEL CASO
Hombre de 35 años de edad que inicia padecimiento actual tres días antes de su ingreso hospitalario con fiebre no cuantificada, tos con expectoración amarillenta, disnea y cianosis peribucal. Veinticuatro horas posteriores al cuadro inicial, se presenta dolor abdominal localizado en cuadrante inferior derecho que inicia de manera súbita e incrementa su intensidad gradualmente. El paciente ingresa con datos clínicos de dificultad respiratoria (taquipnea, cianosis peribucal), la oximetría de pulso marca saturación de 50% spO2, y datos sugestivos de broncoespasmo (sibilancias, disnea progresiva) por lo que se maneja con oxígeno suplementario y micronebulizaciones con salbutamol, budesonida y solución salina.
Cuarenta y ocho horas después ingresa a la Unidad de Cuidados Intensivos con deterioro respiratorio, saturando a 88% a pesar de la administración de oxígeno y una presión arterial de oxígeno (paO2) de 70% en la gasometría, por lo que se decide intubación y manejo con ventilación mecánica asistida (VMA).
Se identifican en el paciente antecedentes de mucopolisacaridosis IV (síndrome de Morquio), retraso psicomotor y estreñimiento constante. Los antecedentes heredofamiliares negados, y antecedentes personales no patológicos sin relevancia.
A la exploración física, hombre de edad aparente menor a la cronológica, con integridad corporal, biotipo mesomórfico, se identifican facies dolorosas, escala de agitación y sedación de Richmond -3, afebril y palidez de tegumentos. La frecuencia cardiaca registró 74 latidos por minuto, frecuencia respiratoria de 16 respiraciones por minuto y presión arterial 110/70 mmHG. Talla de 130 cm, peso 59 kg e IMC 34.9 kg/m2.
Se identifican pupilas simétricas y reactivas a la luz, con opacidad de la córnea, cara redondeada y mandíbula ensanchada; cuenta con cánula endotraqueal con ventilación asistida, saturación de oxígeno de 94% y con fracción de oxígeno inspirada (FIO2) de 70%. Cuello corto y tórax asimétrico, la caja torácica protruye sobre nivel de esternón (pectus carinatum), murmullo vesicular presente, estertores crepitantes bilaterales y ruidos cardiacos rítmicos. Se percibe abdomen distendido, peristálsis aumentada, con facies dolorosas a la palpación de cuadrante inferior derecho y datos de irritación peritoneal. Registró gasto urinario por arriba de 0.5 cc/kg/hr. Las extremidades superiores e inferiores íntegras hipotróficas e hipotónicas y miembros inferiores con desviación de las rodillas hacia adentro (genu valgum).
De acuerdo a las manifestaciones clínicas identificadas y por antecedentes relevantes se realizan estudios de laboratorio, los cuales mostraron lo siguiente: anemia normocítica y normocrómica por hemoglobina (Hb) de 7.5 gr/dL; acidosis respiratoria crónica agudizada no compensada; gasometría venosa: pH 6.9, PCO2 111 mm/Hg, PO2 70 mm/Hg, HCO3 24.1 mmol/L, lactato 0.3 mmol/L; hipotiroidismo primario; perfil tiroideo: T3 .4 ng/dL T3 libre: .7 ng/dL T4: 6.9μg/dL T4 libre 1 ng/dL y TSH 411.4 mU/ml.
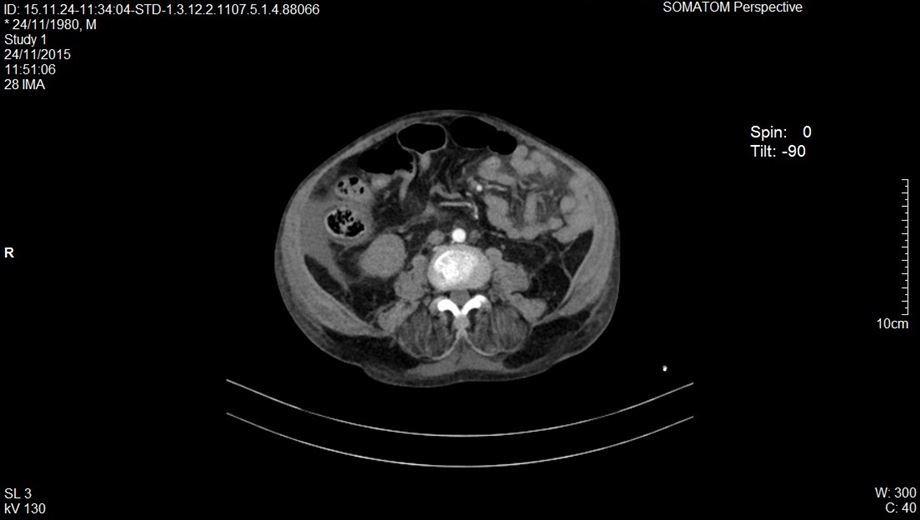
Se decide realizar una tomografía en fase simple y contrastada de tórax y abdomen, reportando en la ventana mediastinal derrame pleural bilateral menor al 20%, tórax restrictivo, en la ventana pulmonar con áreas de consolidación y patrón en vidrio despulido bilateral (figuras 1 y 2). El abdomen presentaba hiperdensidad en ambos riñones compatibles con nefrolitiasis (figura 3). Se identificó líquido libre por la corredera parietocólica derecha hasta formar colección mayor en fosa iliaca ipsilateral (figura 4).
Figura 1. Tomografía computarizada donde se aprecia ventana mediastinal en fase constrastada corte coronal.
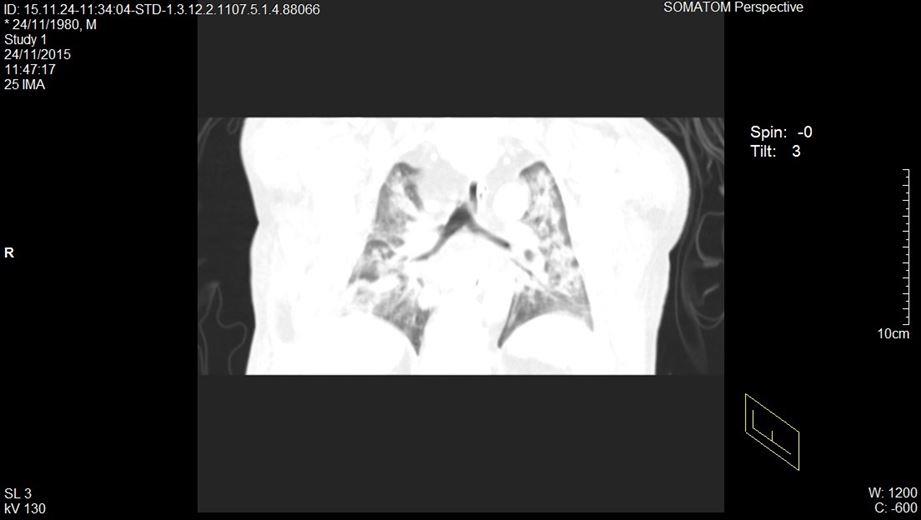
Figura 2. Tomografía computarizada donde se aprecia ventana mediastal en fase contrastada corte axial.
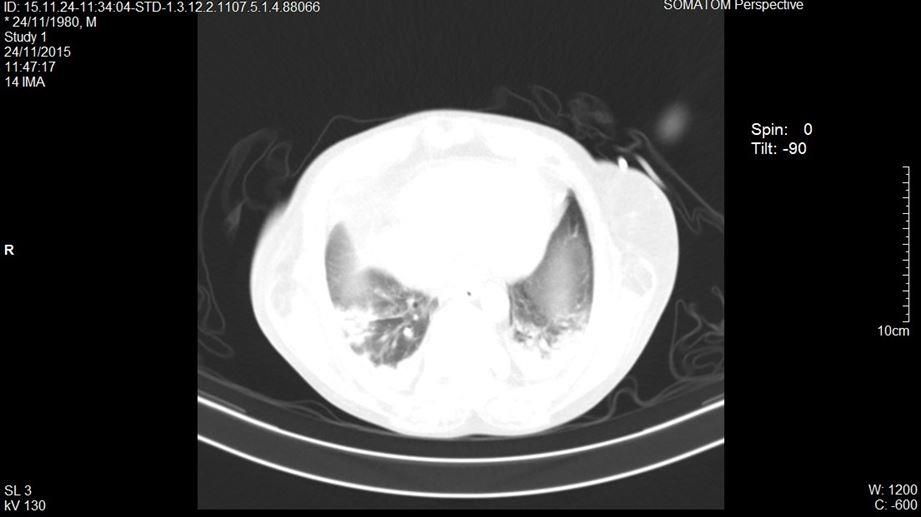
Figura 3. Nefrolitiasis riñón derecho
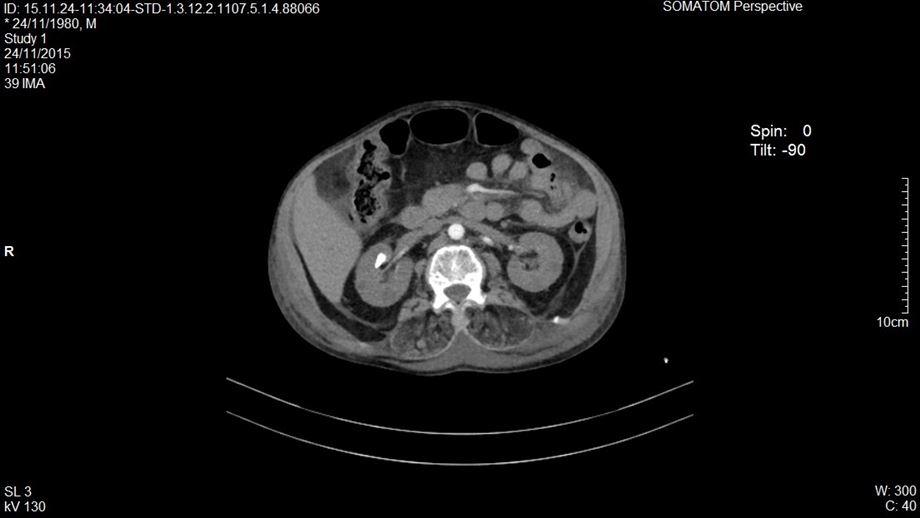
Figura 4. Tomografía computarizada abdominal en fase constrastada corte axial
Se realiza un ecocardiograma, el cual reporta: miocardiopatía crónica metabólica mixta endocrina por hipotiroidismo, enfermedad por depósito infiltrativo, cardiomiopatía restrictiva, crecimiento biauricular secundario, insuficiencia valvular aortica mínima, derrame pericárdico mínimo sin repercusión hemodinámica de importancia, ventrículo izquierdo con disfunción diastólica moderada grado II, y fracción de eyección del ventrículo izquierdo (FEVI) de 55%.
Se establecen los diagnósticos de sepsis, abdomen agudo, neumonía por focos múltiples, hipotiroidismo primario y síndrome anémico. Se continúa el manejo con micronebulizaciones, se inicia tratamiento empírico antimicrobiano para la neumonía, analgésicos y antiespasmódicos para cuadro abdominal, y reemplazo hormonal con levotiroxina por patología tiroidea; se transfunde para estabilización hemodinámica llegando a Hb 12.5 gr/dL, se solicita valoración de cirugía general.
Debido al pronóstico desfavorable por comorbilidades y expectativa de vida, de acuerdo a historia natural de la enfermedad de base, no es intervenido quirúrgicamente. En el transcurso de los días intrahospitalarios y pese al manejo riguroso de acuerdo a guías vigentes, el paciente continua su estado crítico manteniendo un estado de acidosis con Ph de 6.93 e hipotiroidismo primario de recién diagnóstico con grave evolución por mixedema, por lo que se indica administrar terapia hormonal endovenosa y presentando datos de falla orgánica múltiple en progresión (cardiovascular: presión arterial media de 66.6 mmHg a pesar de manejo con aminas; Respiratorio: con VMA por más de dos días, presión positiva al final de la espiración (PEEP) de 8, y síndrome de distres respiratorio agudo por disminución de la relación paO2 entre FiO2 de 155; Renal: oliguria con diuresis de 0.015 cc/kg/hr; hematológico: trombocitopenia de 66,000/mm3 y leucocitosis de 20. 96/mm3 mil; metabólico: hiperglucemia de 385.48 mg/dL).
Se utilizaron medidas de apoyo para corregir hipotensión, hipoxia y oxigenación; se implementaron cambios en antibioterapia y soporte de la patogénesis de la disfunción multiorgánica; sin embargo, por mala evolución del cuadro clínico, el paciente fallece por insuficiencia respiratoria aguda derivada de falla orgánica múltiple secundaria a sepsis.
DISCUSIÓN
Las MPS son parte de las denominadas enfermedades lisosomales. El depósito de GAGS depende del déficit enzimático. El síndrome de Morquio (Mucopolisacaridosis tipo IV), es una enfermedad de depósito lisosómico, de herencia autosómica recesiva. El defecto bioquímico de éste síndrome se encuentra en la actividad deficiente de la N- acetil- galactosamina-6- sulfatasa, enzima encargada de los componentes de la galactose-6- sulfato del sulfato de keratán (10,13).
Las manifestaciones clínicas y comorbilidades asociadas al síndrome de Morquio son secundarias a los depósitos de GAGS presentes en las células de huesos, cartílagos, tendones, córneas, la piel, el tejido conectivo y el tejido hematopoyético.
El síndrome del Morquio es uno de los desórdenes de mucopolisacáridos más raros en el mundo. Las diversas bibliografías sobre esta enfermedad difieren en su incidencia general.
En este caso, el paciente presentó diferentes comorbilidades asociadas al cuadro clínico esperado del síndrome de Morquio, y descritas en otros artículos que revisan esta enfermedad, que incluyen opacidad corneal, problemas cardiacos, y alteraciones esqueléticas (7,13); sin embargo, en los casos existentes no hay asociación o algún reporte de un caso de síndrome de Morquio con hipotiroidismo.
Los pacientes con síndrome de Morquio presentan obstrucción moderada de la vía aérea superior, obstrucción leve de la vía aérea inferior y grave compromiso restrictivo. Esto puede asociarse a que generalmente los pacientes tienen rinorrea nasal crónica o persistente, además de secreciones de la vía aérea superior e inferior, explicado por obstrucción mecánica por infiltración de la mucosa, por un proceso inflamatorio persistente o por la calidad de las secreciones desde las glándulas de la mucosa y submucosa, en donde los principales GAGS son dermatán y keratán sulfato. La restricción pulmonar es condicionada por alteraciones de la caja torácica por compromiso esquelético severo, ya que debido a que el crecimiento de la espina dorsal es afectada, el esternón es forzado a doblarse hacia afuera en una curvatura redondeada o en forma de pico. El pecho es en forma de campana y las costillas que están fijas en una posición horizontal causan la restricción eficiente respiratoria.
Las personas con síndrome de Morquio, de acuerdo a las alteraciones explicadas anteriormente, y sumado el compromiso neurológico, los predisponen a infecciones y neumonías recurrentes y la restricción en el pecho los hace menos capaces de confrontar una infección de pulmón (14).
La falta de información propicia desconocimiento de la patología, lo que limita el manejo multidisciplinario, propuesto en diversos artículos, que este paciente requiere.
CONCLUSIÓN
El síndrome del Morquio es uno de los desórdenes de mucopolisacáridos más raros en el mundo, es por eso que la información disponible es insuficiente y desactualizada. Siendo una enfermedad progresiva, cuyos síntomas se presentan en la medida que el acúmulo de mucopolisacáridos afecta diversos sistemas, su manejo es multidisciplinario, debido al amplio compromiso sistémico involucrado. La mortalidad y morbilidades asociadas son elevadas, por lo tanto la intervención oportuna es la clave para alcanzar una buena calidad de vida. Se espera que el antecedente de este caso con una comorbilidad no documentada previamente sirva como precedente para el estudio de casos similares.
REFERENCIAS BIBLIOGRÁFICAS
1. Neufeld EF, Münzer J. Las bases metabólicas y moleculares de la enfermedad hereditaria, Scriver C, Beaudet AL, Valle D, Sly W (Eds), McGraw-Hill, Nueva York 2001. p.3421.
2. Wenger DA, Coppola S, Liu SL. Insights en el diagnóstico y tratamiento de las enfermedades de almacenamiento lisosomal. Arco Neurol 2003; 60: 322.
3. Baker, E, Guo XH, Orsborn AM, et al. El síndrome de Morquio A (IVA mucopolisacaridosis) mapas de genes a 16q24.3. Am J HumGenet 1993; 52:96.
4. Northover H, Cowie RA, Wraith JE. Mucopolysacchariosis type IV A (Morquio Syndrome): A clinical review. J InherMetabDis 1996; 19: 357-365
5. Maroteaux P, Stanescu V, Stanescu R, Kresse H, Hors-Cayla MC. Heterogeneite des formes frustes de la maladie de Morquio. Arch Fr Pediatr 982;39:761-5.
6. Mikles M, Stanton RP. Una revisión del síndrome de Morquio. Am J Orthop (Belle Mead NJ) 1997; 26: 533.
7. Nelson J, Broadhead D, Mossman J. hallazgos clínicos en 12 pacientes con MPS IV A (enfermedad de Morquio). Otra prueba para la heterogeneidad. Parte I: Los hallazgos clínicos y bioquímicos. ClinGenet 1988; 33: 111.
8. Holzgreve W, Grobe H, von Figura K, et al. El síndrome de Morquio: hallazgos clínicos en 11 pacientes con MPS IVA y 2 pacientes con MPS IVB. HumGenet 1981; 57: 360.
9. Wappner RS. Trastornos de almacenamiento lisosomal. En: Pediatría de Oski. Principios y Práctica, 4ª ed, McMillan JA, Feigin RD, DeAngelis C, Jones MD (Eds), Lippincott, Williams &Wilkins, Philadelphia 2006. p.2199.
10. Suárez-Obando F, Zarante-Montoya I. Aspectos clínicos y manejo íntegral del síndrome de Morquio. Universitas Médica 2007, VOL.48
11. Wappner RS. Trastornos de almacenamiento lisosomal. En: Pediatría de Oski. Principios y Práctica, 4ª ed, McMillan JA, Feigin RD, DeAngelis C, Jones MD (Eds), Lippincott, Williams &Wilkins, Philadelphia 2006. p.2199.
12. Neufeld E, Muezner J. The mucopolysaccharidoses. In: The metabolic and molecular bases of inherited disease. Eightedition. New Yorl: McGraw-Hill Professional; 2000. Pages:2300-2303.
13. Salinas H, Preisler J, Astudillo J, Cerda S, Castillo, Fernández F, et al. Síndrome de morquio (mucopolisacaridosis tipo iv) y embarazo.REV CHIL OBSTET GINECOL 2005; 70(6): 400-403.
14. Hernández J. Compromiso respiratorio en mucopolisacaridosis. NeumolPediatr 2013; 8 (1): 27-33.