KABUKI MAKE UP SYNDROME. ANALYSIS OF A CASE
Cristóbal M. Valdez Geraldo*, Sayra García Arias**, Blanca M. Valdez Geraldo***, O. García-Torres***
RESUMEN
El síndrome Kabuki es una enfermedad pediátrica extremadamente rara, descrita por dos médicos japoneses Niikawa y Kuroki , llamada así debido a la semejanza que estos pacientes tienen con los actores maquillados de la modalidad del teatro japonés. Cuenta con manifestaciones clínicas pleomórficas, entre las cuales resaltan los hallazgos faciales característicos, entre otros, el alargamiento pronunciado de las fisuras palpebrales, la eversión en el tercio externo del borde superior de ambos párpados inferiores, redondeamiento y aplanamiento de la punta de la nariz, puente nasal ancho, orejas grandes y prominentes con implantación baja, arco palatino elevado con anomalías dentales; anormalidades esqueléticas, talla baja, retraso mental y del crecimiento. Incluyendo alteraciones inmunológicas y hematológicas del tipo de la púrpura trombocitopenica. Este es el segundo caso reportado en México.
Palabras clave: Dismorfia facial, síndrome de Kabuki, eversión del tercio externo de párpados inferiores, retraso mental.
ABSTRACT
Kabuki syndrome is a pediatric disease extremely rare, described by two Japanese physicians Niikawa and Kuroki, so called, by extraordinary resemblance that these patients have with actors makeup of the modality of Japanese theater. It has pleomorphic clinical manifestations, which include its importance by the characteristic facial features, highlighting among others, the pronounced elongation of palpebral fissures, everted lateral 1/3 of the lower lids, and flattened and rounded tip of the nose, nasal bridge wide, large and prominent ears with low implantation, high palatal arch and dental anomalies; skeletal abnormalities, short stature, mental retardation and growth delay 2 Including immune alterations like thrombocytopenic purpura. This is a second case reported in México.
Key words: Facial Dysmorphy, Kabuki, everted lateral 1/3 lower lids, mental retardation.
INTRODUCCIÓN
El primer caso del síndrome Kabuki fue reportado en Japón por Niikawa y Kuroki (1,2) quienes describieron las características morfológicas faciales típicas: talla baja, anormalidades esqueléticas, problemas cardiacos, auditivos y retraso mental. Su frecuencia en Japón es de 1/32,000 nacidos vivos. La microdelecion cromosómica que no siempre es demostrable en el cariotipo es la que más se ha encontrado, así como la duplicación del cromosoma 8,22q11.2, 1q32-q41.(3,4,5) Los datos clínicos esenciales son alteraciones morfológicas faciales típicas como eversión palpebral inferior, fisuras largas, cejas arqueadas, pestañas grandes, escleróticas azules, punta nasal plana, paladar fisurado, arqueado y/o labio leporino, orejas malformadas, fístula preauricular, dentadura anormal, retraso en el crecimiento, estatura baja, anormalidades esqueléticas, dermatoglíficas y retraso mental. La mayoría de los pacientes también suelen tener defectos cardiacos, hematológicos y problemas de la audición.(6) Se asocia a menudo con alteraciones inmunológicas y hematológicas, de las cuales la más frecuente es la púrpura trombocitopénica idiopática.(7) Aunque hay un subregistro de casos diagnosticados por la gama de síntomas que se mencionan en los artículos hasta ahora publicados, se han informado 300 pacientes, 130 japoneses y 170 en el resto del mundo.(7,8,9,10,11,12) En México solo existe el reporte de un caso.(13)
No existe una prueba específica para esta enfermedad y el diagnóstico se realiza con criterios clínicos en base a las dismorfias.(2)
PRESENTACIÓN DEL CASO
Masculino de 8 años de edad, que fue estudiado debido a la recurrencia de púrpura trombocitopenica idiopática, la cual ya había padecido hacía un año. Producto de la tercera gestación, de unión libre de padres no consanguíneos, padre policía municipal de 35 años de edad, madre de 27 años, mucama de hotel, con amenaza de aborto a las 16 semanas de gestación, pretérmino de 35 semanas, nació por parto vaginal con peso de 2,700 grs. Cursó con neumonía intrauterina, ameritando estancia hospitalaria una semana en UCI neonatal.
Alimentado al seno materno hasta los 8 meses de edad, desarrollo psicomotriz con rezago, logrando sostén cefálico hasta los 6 meses, sedestación a los 11 meses y deambulación sin ayuda hasta los 2 años y medio de edad.
Comenzó a pronunciar disílabos a los 2 años y medio de edad y frases a los 4 años. Inició sintomatología un año antes del diagnóstico, padecimiento que cursó con equimosis y petequias en brazos y piernas. Recibió tratamiento con prednisona por una semana, permaneciendo asintomático 7 días, al cabo de los cuales presenta recaída con epistaxis durante 3 días, las equimosis y petequias ameritaron su primer ingreso al hospital, siendo dado de alta por mejoría 7 días después.
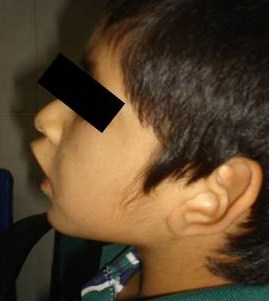
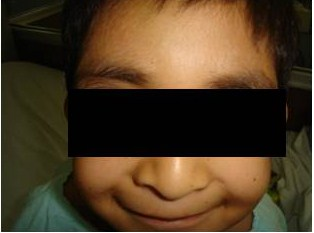
Un año después, presentó nuevamente equimosis, petequias y epistaxis y trombocitopenia de 21,000 plaquetas, ameritando un segundo ingreso hospitalario. Veinticuatro horas después de ser tratado con inmunoglobulina G IV, y prednisona oral, se encontró Hemoglobina de 13.2, Hematocrito de 37.2, 11 mil plaquetas. Al examen físico, talla de 118 cm., peso 20 Kg. y perímetro cefálico de 49 cm. La antropometría por debajo del percentil 10. Sus rasgos faciales denotaban semejanza asiática con fisuras palpebrales alargadas, oblicuas, hipertelorismo, filtrum largo, cejas arqueadas, eversión del tercio externo de ambos párpados inferiores (figura 1), orejas prominentes antevertidas y de implantación baja (figura 2).
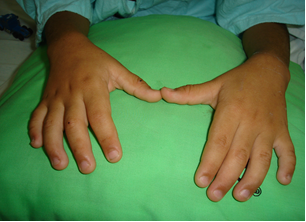
Su habitus corporal adelgazado, con manos pequeñas, con campodactilia y clinodactilia, persistencia de almohadillado fetal de pulpejos, en las manos en las regiones tenar e hipotenar, dermatoglifos con predominio de asa cubitales semejando pliegue simiano (figura 3) con laxitud ligamentaría en pulgares y articulación del carpo. Había paladar arqueado y fisura palatina. Sus niveles de inmunoglobulinas fueron normales.
Una TAC craneal simple fue normal a excepción de una variedad de Cisterna Magna agrandada, su electroencefalograma mostró datos de inmadurez bioeléctrica debido a ausencia de gradiente anteroposterior por falta de integración del ritmo alfa occipital. Su evaluación cognitiva mediante la prueba estandarizada del Neurodesarrollo para escolares Brigance Assessment of Basic Skills (ABS-R) denotó una puntuación por debajo de la media para su edad. El cariotipo fue normal sin evidencias de delección cromosómica.Por sus características morfológicas se concluyó que era un síndrome de Kabuki.
Actualmente el paciente esta siendo tratado por sus alteraciones hematológicas, inmunológicas y neurológicas en el Hospital del IMSS No 1 de la Paz que es la clínica de concentración en la zona. Fue referido a genética para su estudio complementario. La vigilancia estrecha neuropsicológica del niño y su desarrollo están a cargo de neurología pediátrica, para evitar mayor deterioro y ayudar a su rehabilitación e integración a su comunidad
DISCUSIÓN
Presentamos este caso en razón de las características clínicas prototípicas encontradas, específicamente las del tipo dismórfico-facial, típicas, semejantes a las observadas en las máscaras de los actores del teatro japonés Kabuki, cuyo vocablo original significa “inexpresivo” y las que se encontraron en nuestro paciente, tales como la oblicuidad palpebral oriental alargada con eversión del tercio externo de los párpados, el aplanamiento nasal, las orejas prominentes y el tatuaje de los bordes palpebrales combinados con el patrón dermatoglífico y de depósito persistente de almohadillas de grasa fetal en manos y pulpejos; así como su asociación con manifestaciones hematológicas, indicativas de alteración inmunológica subyacente.
A este respecto, el síndrome se caracteriza por alteración de la inmunidad humoral, de las células B, resultando en susceptibilidad aumentada a infecciones respiratorias. Ochenta por ciento de los pacientes presentan disminución de los niveles de Inmunoglobina A, 40% Inmunoglobina G disminuida, siendo normal los valores de Inmunoglobina M y la función de la inmunidad celular de células T. (9)
Asimismo suelen tener enfermedades autoinmunes como la púrpura trombocitopenica como en nuestro paciente y otras afecciones tales como el vitiligo, tiroiditis y anemia hemolítica como resultado de pérdida en la autorregulación. (7,8) No existe un patrón genotípico característico y se ignora la causa del padecimiento, aunque en algunas comunidades de Japón se encontró herencia autosómica dominante de expresión variable. (15)Los primeros casos descritos posiblemente estén entre la tercera y cuarta décadas de la vida. En Latinoamérica solo hay cuatro casos reportados, uno en Brasil,(10) otro en Haití,(11) uno en Ecuador,(12) y uno más en México.(13) En España hay 18 casos documentados.(14)
CONCLUSIÓN
En particular nuestro caso, el segundo publicado en México, sin transmisión hereditaria aparente, ni consanguinidad parental, actualmente se encuentra a cargo del servicio de hematología pediátrica para vigilancia de su afección plaquetaria, además del servicio de Alergia e Inmunología debido a sus alteraciones inmunitarias y por el servicio de Neuropediatría y los Servicios de Educación Especial de la Secretaría de Educación Pública para seguimiento e integración escolar y en apoyo de su desarrollo cognitivo y revertir tentativamente sus alteraciones de aprendizaje.
REFERENCIAS BIBLIOGRÁFICAS
1 Kuroki Y, Susuki Y,Chio H, Hata A, Matsui I: A new malformation Syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr 1981;99:570-3.
2 Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up síndrome: a síndrome of mental retardation, unusual fascies, large and protruding ears and postnatal growth deficiency. J Pediatr 1981;99: 565-9.
3 Matsumoto N, Niikawa N. Kabuki makeup syndrome: a review, Am J Med Genetic 2003; 117: 57/65.
4 Milunsky JM, Huang XL. Unmasking Kabuki syndrome: chromosome 8p22–8p23.1 duplication revealed by comparative genomic hybridization and BAC-FISH. Clin Genet 2003: 64: 509–516
5 Tsukahara M, Kuroki Y, Imaizumi K, Miyazawa Y, Matsuo K. Dominant inheritance of Kabuki make-up syndrome. Am J Med Genet 1997:73:19–23.
6 Adam MP, Hudgins L. Kabuki syndrome: A review. Clin Genet 2004;67:209-19.
7 Watanabe T, Myakawa M, Satoh M, Abe T, Oda Y. Kabuki make-up syndrome associated with chronic idiopathic thrombocytopenic purpura. Acta Paediatr Jpn 1994 ;36:727-9.
8 Ming JE, Rusell KL, McDonald-McGinn DM, Zackai EH. Autoinmune disorders in Kabuki syndrome. Am J Med Genet 2005;132:260-62.
9 De Souza JC, Ribeiro TC, Ribeiro RC. Kabuki make-up syndrome. J Pediatr (Rio J) 1996;72:341-4.
10 Milunsky JM, Cheney SM. Kabuki syndrome in a Haitian patient. Am J Med Genet 2001;100:172-4.
11 Guzmán AAM, Tumbaco R, Jaramillo L. Kabuki syndrome. Rev Ecuat Neurol 2004; 13:31-2.
12 Aviña-Fierro J, Pérez-Ornelas N. Síndrome de Kabuki: Informe de un caso. Acta Pediatrica Mex 2006; 27(6): 349-51.
13 Pascual Cl, Pascual PCI, Velazquez FR, Palencia R.Kabuki make-up syndrome. A report of Spanish cases. Rev Neurol 2005; 40:473-8.
14 Tsukahara M, Kuroki Y, Imaisumi K, Miyazawa Y, Matsuo K. Dominant Inheritance of kabuki make-up síndrome. Am J Med Gen 1997;73:19-23.
* Neurólogo Pediatra. Hospital General de Zona. Instituto Mexicano del Seguro Social (IMSS) “Enrique Von Borstel Labastida”, La Paz, BCS.
**Neonatóloga Pediatra. Hospital General de Zona 1. Instituto Mexicano del Seguro Social (IMSS) “Enrique Von Borstel Labastida”, La Paz, BCS.
***Médico Familiar. Hospital General de Subzona 6 IMSS, San José del Cabo, BCS.
Correspondencia: Dr. Cristóbal Medardo Valdez Geraldo. Durango 1725 Colonia Guerrero, La Paz, Baja California Sur, México C.P. 23020 Tels: (612) 1253109. Correo electrónico: pedimss@yahoo.com.mx; neuro54@hotmail.com